
Abstract
Singe crystals of a new high-temperature polymorph of Na2Ca6Si4O15 have been obtained from solid state reactions performed at 1300 °C. The basic crystallographic data of this so-called β-phase at ambient conditions are as follows: space group P1c1, a = 9.0112(5) Å, b = 7.3171(5) Å, c = 10.9723(6) Å, β = 107.720(14)°, V = 689.14(7) Å3, Z = 2. The crystals showed twinning by reticular merohedry (mimicking an orthorhombic C-centred unit cell) which was accounted for during data processing and structure solution. Structure determination was accomplished by direct methods. Least-squares refinements resulted in a residual of R(|F|) = 0.043 for 5811 observed reflections with I > 2σ(I). From a structural point of view β-Na2Ca6Si4O15 can be attributed to the group of mixed-anion silicates containing [Si2O7]-dimers as well as isolated [SiO4]-tetrahedra in the ratio 1:2, i.e. more precisely the formula can be written as Na2Ca6[SiO4]2[Si2O7]. The tetrahedral groups are arranged in layers parallel to (100). Sodium and calcium cations are located between the silicate anions for charge compensation and are coordinated by six to eight nearest oxygen ligands. Alternatively, the structure can be described as a mixed tetrahedral-octahedral framework based on kröhnkite-type [Ca(SiO4)2O2]-chains in which the CaO6-octahedra are corner-linked to bridging SiO4-tetrahedra. The infinite chains are running parallel to [001] and are concentrated in layers parallel to (010). Adjacent layers are shifted relative to each other by an amount of +δ or −δ along a*. Consequently, a …ABABAB… stacking sequence is created. A detailed comparison with related structures such as α-Na2Ca6Si4O15 and other A2B6Si4O15 representatives including topological as well as group theoretical aspects is presented. There are strong indications that monoclinic Na2Ca3Si2O8 mentioned in earlier studies is actually misinterpreted β-Na2Ca6Si4O15. In addition to the detailed crystallographic analysis of the previously unknown compound our results will also help to improve the interpretation of the phase relationships between the compounds in the ternary system Na2O-CaO-SiO2 which are of interest for several applications related to the field of applied mineralogy and materials science.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Among the silicates containing alkali and alkaline earth cations the compounds belonging to the system Na2O-CaO-SiO2 have attracted the most attention. This is not really surprising, because the silica rich part of this system is of fundamental importance for manufacturing of so-called soda-lime-glasses which are the major commercial types used for the production of hollow and flat glasses such as containers or windows, respectively (Varshneya 1994; Shelby 2009).
Since the landmark paper of Morey and Bowen (1925), several subsequent studies focused on the phase relationships as well as melting temperatures and melting behaviour of sodium calcium silicates (Thilo 1951; Segnit 1953; Shahid and Glasser 1970, 1971; Moir and Glasser 1974; Garza Montoya et al. 2003; Zhang et al. 2011; Knowles and Thompson 2014). However, especially the earlier investigations frequently lacked precise knowledge of the chemical formulas of the proposed phases. Therefore, many of the compounds disclosed in these phase equilibrium studies had to be later characterized by diffraction methods not only for the determination of their crystal structures but also for proving their suggested chemical compositions, i.e. structure analysis played the role of a convenient analytical probe. In the meantime crystal structure investigations have been performed for the following compounds: Na2CaSiO4 (Dollase and Ross 1991), Na2Ca3Si3O10 (Treushnikov et al. 1971), Na4CaSi3O9 (Fischer and Tillmanns 1984), α-Na2Ca2Si3O9 (Ohsato et al. 1990), β-Na2Ca2Si3O9 (Fischer and Tillmanns 1987), Na2CaSi2O6 (Ohsato et al. 1985), Na2Ca3Si6O16 (Ihara et al. 1984; Kahlenberg et al. 2010), Na2Ca2Si2O7 (Kahlenberg and Hösch 2002) and Na2Ca6Si4O15 (Armbruster and Röthlisberger 1990).
Due to their large variety in chemical composition the group of sodium-calcium-silicates is an excellent pool for the crystallographer to study the influence of the ratios between tetrahedrally and non-tetrahedrally coordinated cations on the topology of the resulting crystal structures. A summary of their structural characteristics can be found in Kahlenberg and Hösch (2002).
However, there is still a large number of ternary compounds in the system Na2O-CaO-SiO2 that have been mentioned in the literature for which no or only basic crystallographic data in terms of unit cell parameters are available. Therefore, to a certain extent suspicion has to be attached to their existence. One of the phases belonging to this group is Na2Ca3Si2O8 or Na2O × 3CaO × 2SiO2. To the best of our knowledge, Maki (1969) was the first who presented some information about this compound. According to his study which aimed on the preparation of new alkali oxide containing Ca2SiO4 analogues, a stable triclinic and a metastable monoclinic modification of Na2Ca3Si2O8 have to be distinguished. The monoclinic form could be prepared at 1300 °C and transformed slowly into a triclinic phase after prolongated annealing for more than 100 h at this temperature. He also presented indexed powder diffraction patterns of both polymorphs having the following sets of lattice parameters: triclinic form: a = 5.47 Å, b = 7.32 Å, c = 9.02 Å, α = 91.37°, β = 107.70, γ = 89.80°; monoclinic form: a = 10.99 Å, b = 7.33 Å, c = 9.01 Å, β = 107.75°. Maki’s efforts to index the powder patterns of both low-symmetry compounds from laboratory data cannot be appreciated enough. His high quality data were later used for creating the entries 00-023-0668 and 00-023-0670, respectively, of the PDF-4 Powder Diffraction File database issued by the International Centre for Diffraction Data (ICDD). These entries in turn were the primary source for phase identification of polycrystalline Na2Ca3Si2O8 in several subsequent papers using diffraction methods (Zhang et al. 2009; Kuo 2014; Jin et al. 2015; Fu and Hu 2016). Unfortunately, however, Maki did not perform a more detailed chemical analysis of his synthesis products.
The surprising lack of knowledge concerning more detailed crystallographic data on Na2Ca3Si2O8 prompted us to include this phase into an ongoing long-time study on the phase relationships and the crystal chemistry of silicates containing alkali as well as alkaline earth oxides. In this publication we present our results indicating that – at least – monoclinic Na2Ca3Si2O8 probably corresponds to a previously unknown high-temperature modification of Na2Ca6Si4O15.
Experimental details
Synthesis
Synthesis experiments of ternary oxides with the molar ratio Na2O:CaO:SiO2 = 1:3:2 were based on mixtures of the following dried educts: Na2CO3 (Alpha Aesar, 99.9 %), CaCO3 (Merck, >99.9 %) and quartz (AlfaAesar, 99.995 %). The starting materials were homogenized for 45 min under ethanol in a planetary mill with 600 r.p.m., dried over night at 60 °C (in order to completely remove the ethanol) and stored in a desiccator. Afterwards, the educts were pressed into pellets (approximately 0.5 g, each having a diameter of 12 mm and a thickness of about 3 mm) and dried again at 300 °C. A single pellet was transferred into an open platinum crucible, placed into a resistance heated furnace and fired from 300 °C with a heating ramp of 50 °C per hour to 1300 °C. The crucible was removed after 44 h and finally quenched in air. Weight loss was determined from weight difference before and after heating.
Assuming a complete disintegration of the carbonates, one would expect a theoretical weight loss of 33.44 %. The corresponding value of our synthesis experiment performed at 1300 °C was 39.51 %, i.e. the experimentally determined loss is much higher. It seems to be likely to attribute the excess weight loss to an evaporation of Na2O, which is known to be the most volatile oxide in this system.
A first inspection of the run-product indicated the presence of single crystals up 100 μm in diameter embedded in a polycrystalline matrix. The crystals could be separated mechanically. First investigations using a petrographic microscope showed that the optical quality of the colorless transparent crystals was only moderate. Except for very small samples most of the crystals exhibited undulose extinction when observed under crossed polarizers.
Chemical analysis
One of the crystals obtained from the synthesis experiment was embedded in epoxy resin and polished with diamond paste (3 and 1 μm grain size) for chemical analysis with an electron microprobe (JEOL JXA 8100 Superprobe) operated in wavelength-dispersive mode. Measurements were performed with an acceleration voltage of 15 kV and a 10 nA beam current. For calibration of sodium, calcium and silicon standard specimens of jadeite, diopside and quartz were used. The intensities obtained were corrected for electron scattering, absorption and fluorescence radiation (so-called ZAF correction). A total number of six spots in various areas of the crystal were analysed. They all yielded a very similar chemical composition giving no indication for a chemical zoning. The average values in weight-% are as follows: Na2O: 9.32(3), CaO: 53.28(2) and SiO2: 37.40(2). After normalization to 15 oxygen atoms almost integer atomic ratios were obtained: Na : Ca : Si : O = 1.92(2) : 6.06(2) : 3.97(1) : 15. This resulted in an idealized chemical formula of Na2Ca6Si4O15 and pointed to the presence of a phase the composition of which is significantly different from Na2Ca3Si2O8 (corresponding to 17.69 % Na2O, 48.01 % CaO and 34.30 % SiO2).
Single-crystal diffraction
Several single crystals of apparently good optical quality were fixed on the top of glass fibres with nail polish and examined on a Stoe IPDS-II imaging-plate diffractometer at room conditions. Many samples, however, showed reflections which were radially elongated making them unsuitable for more detailed data collections. Therefore, we focused on one of those that were of better diffraction quality. At a first glance, the diffraction pattern could be indexed with an orthorhombic C-centred unit cell. The corresponding lattice parameters had values of a OR = 34.332(2) Å, b OR = 10.9723(6) Å and c OR = 7.3171(5) Å. However, a more detailed inspection using precession-type reconstructions of reciprocal space revealed systematic absences that were not compatible with any orthorhombic space group. Actually, the diffraction pattern could be explained as the result of twinning by reticular merohedry of two monoclinic primitive individuals mimicking orthorhombic symmetry. Principally, two classes of reflections could be distinguished containing (i) contributions from only one domain each (complete separation) and (ii) contributions from both domains (complete overlap). The program X-Area (Stoe & Cie GmbH 2005) of the STOE diffractometer software package was employed to isolate the diffraction spots coming from the two different orientations of the domains. The diffraction peaks were indexed independently and the superimposed diffraction patterns were integrated simultaneously. Integration was repeated twice (with and without overlap check) for each twin domain. After transformation to monoclinic second setting the following lattice parameters were obtained for the domain I with the higher mean intensity: a = 9.0112(5) Å, b = 7.3171(5) Å, c = 10.9723(6) Å, β = 107.720(14)°. Further data reduction included Lorentz and polarization corrections. No absorption correction was applied. Systematic absences pointed to space groups P1c1 or P12/c1. Structure solution was performed by direct methods (program SIR2004, Burla et al. 2005) using the data set containing the non-overlapping reflections from domain I only. However, only in the acentric space group P1c1 a crystal chemical reasonable model showing SiO4-tetrahedra and additional Na/Ca cation sites was obtained. For the least-squares refinement of this initial model (program SHELX-97 (Sheldrick 1997) embedded in the WinGX software suite (Farrugia 1999)) the data set containing the overlapping and non-overlapping reflections from both domains (HKLF 5 command) was used. The volume fractions of the two twin domains refined to values of 70.9(2):29.1(2) respectively. Neutral atom scattering coefficients and anomalous dispersion corrections were taken from the International Tables for Crystallography, Volume C (Wilson 1995). A critical check of the structure and its space group including the MISSYM algorithm implemented in the program PLATON (Spek 2009) did not reveal any indication that a wrong or unnecessarily low space group symmetry had been chosen. The final computations using anisotropic displacement parameters for all atoms as well as site occupancy refinements for the Na/Ca-positions resulted in a residual of R1 = 0.0430 for 246 parameters for all observed reflections (see Table 1). The difference electron density map turned out to be featureless, with maxima and minima of 1.14 and −1.13 e∙Å−3. The chemical formula derived from the structure analysis corresponds to Na2Ca6Si4O15 and is in excellent agreement with the results from the electron microprobe analysis. The optimized atomic coordinates, anisotropic displacement parameters as well as selected interatomic distances and angles are given in Tables 2, 3 and 4, respectively. Figures showing structural details were prepared using the program ATOMS6.4 (Dowty 2011).
Results
Description of the crystal structure
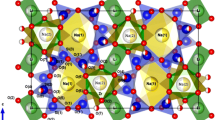
The present phase represents a second polymorph of Na2Ca6Si4O15. For sake of clarity the already known modification first described by Armbruster and Röthlisberger (1990) and the new form will be designated α- and β-Na2Ca6Si4O15, respectively. The crystal structure of β-Na2Ca6Si4O15 belongs to the group of mixed-anion silicates. It is built from isolated [SiO4]-tetrahedra as well as [Si2O7]-units in the ratio of 2:1, i.e. more precisely the formula could be written as Na2Ca6[SiO4]2[Si2O7]. As shown in Fig. 1, the tetrahedral groups are arranged in layers parallel to (100). Sodium and calcium cations are located between the [SiO4]-groups and [Si2O7]-dimers for charge compensation. In more detail, a total of eight symmetrically independent non-tetrahedral positions can be distinguished. With regard to the distribution of the Si-O bond lengths of the anion groups a pronounced distortion of the tetrahedra can be noticed. The spread of the Si-O distances ranges from 1.562–1.655 Å. Nevertheless, the observed values are in accordance with accepted values for silicate structures (Liebau 1985). The distortion is also reflected in the O-Si-O angles ranging from 101.3° to 117.8°. The average <O-Si-O> angles are, however, very close to those observed for an undistorted tetrahedron. The distortions can be expressed numerically via the quadratic elongations λ and the angle variances σ2 (Robinson et al. 1971). The values of these parameters for the four crystallographically independent [SiO4]-units are as follows: Si(1): λ = 1.003 and σ 2 = 9.61, Si(2): λ = 1.005 and σ 2 = 20.96, Si(3): λ = 1.006 and σ 2 = 24.32 and Si(4): λ = 1.006 and σ 2 = 23.19, i.e. the tetrahedra around Si(3) and Si(4) forming the dimer are slightly more distorted than the two isolated [SiO4]-moieties around Si(1) and Si(2). Apart from O(10) all oxygen atoms in Na2Ca6Si4O15 are non-bridging. The Si(4)-O(10)-Si(3) angle has a value of 157.5(4)° indicating that the [Si2O7]-dimer is not straight but bended. The torsion angles <(O(1)-Si(4)-Si(3)-O(14)) = −19.3(9)°, <(O(6)-Si(4)-Si(3)-O(4)) = −21.8(9)° and <(O(15)-Si(4)-Si(3)-O(9)) = −19.1(9)° show that the conformation of the dimer can be considered as eclipsed. Initial site population refinements of the sodium and calcium ions on the non-tetrahedrally coordinated sites revealed that five out of eight positions are exclusively occupied by Ca ions (Ca(2), Ca(4), Ca(5), Ca(6) and Ca(7)), whereas the Na(8) position is a pure Na-site. The Na/Ca-distributions on the remaining two sites were optimized using the constraint that both positions are fully occupied. The derived populations (resulting in a charge balanced composition) are as follows: M(1): 84(2)% Ca/16(2)% Na and M(3): 16(2)% Ca/84(2)% Na. The positions Ca(2) and Ca(4) are coordinated by six nearest oxygen neighbors in form of distorted octahedra. Each octahedron shares common corners with four adjacent insular tetrahedra and two different [Si2O7]-dimers. The coordination spheres of the remaining Na/Ca-sites are much more irregular involving 7–8 oxygen ligands.

Projection of the whole crystal structure of β-Na2Ca6Si4O15 parallel to [010]. Light and dark blue spheres correspond to pure sodium and cacium sites, respectively. Medium blue spheres represent positions with mixed Ca/Na populations. Oxygen atoms are shown as small grey spheres
Alternatively, the structure can be described as a mixed tetrahedral-octahedral framework (see Fig. 2a). Within this network, infinite chains running parallel to [001] can be identified, in which the [CaO6]-octahedra are corner-linked to bridging [SiO4]-tetrahedra (see Fig. 3a). These so-called “kröhnkite-type” chains (named after the mineral kröhnkite, Na2Cu(SO4)2 × 2H2O) are well known fundamental building blocks of structural inorganic chemistry (Kolitsch and Fleck 2006 and references cited therein). In the present compound, these chains are located in 4.55 Å wide layers running parallel to (010). Adjacent sheets are shifted relative to each other by an amount of +δ or −δ. Consequently, a …ABABAB… stacking sequence is created, where chains of neighboring layers are linked by corner sharing of common oxygen atoms (see Fig. 2a). The resulting three-dimensional network contains tunnel-like cavities in which the remaining more irregularly coordinated Na- and Ca-ions are incorporated. Principally, the α-form first described by Armbruster and Röthlisberger (1990) shows the same structure motifs. However, in this polymorph a four layer stacking …ABCDABCD… of the chains is observed. The series of relative shifts is…+δ,+δ,−δ,−δ,+δ,+δ,… (see Fig. 2b)

Mixed tetrahedral-octahedral frameworks of (a) β-Na2Ca6Si4O15 (b) α-Na2Ca6Si4O15 and (c) Na2Ba6Si4O15 in projections parallel to the directions of the kröhnkite-type chains

Projections of a single kröhnkite-type chain in (a) β-Na2Ca6Si4O15 and (b) Na2Ba6Si4O15 perpendicular to the mean plane of the chain
Comparison with related structures
Kröhnkite-type chains have been already shown to be major constituents of several silicates with the general composition A2B6Si4O15. Apart from both Na2Ca6Si4O15 polymorphs, K2Ca6Si4O15 (Arroyabe et al. 2009) and Na2Ba6Si4O15 (Tamazyan et al. 1987) also belong to this family of crystal structures. Table 5 presents a summary of the basic crystallographic data of these materials. It is obvious, that the translation period of the kröhnkite-type chains of about 5.5-5.6 Å is reflected in one of the lattice parameters of each phase. Furthermore, the comparisons of (i) the unit cell metrics as well as (ii) the graphical representations of β-Na2Ca6Si4O15 and Na2Ba6Si4O15 given in Figs. 2a/c and 3a/b point to a closer relationship between both compounds. In order to characterize the four mixed tetrahedral-octahedral frameworks in more detail a topological analysis has been performed using the program TOPOS4.0 (Blatov 2012). Therefore, the crystal structures have been described by graphs composed of the vertices (sites of the tetrahedrally and octahedrally coordinated cations as well as O anions) and edges (bonds) between them. The nodes of the graph can be classified according to their coordination sequences {N k }. They represent a set of integers {N k } (k = 1,..,n), where N k is the number of sites in the kth coordination sphere of the respective atom that has been selected to be the central one. The corresponding values for the relevant symmetrically independent cation sites up to n = 10 (without the oxygen nodes) are listed in Table 6. Furthermore, the corresponding extended point symbols listing all shortest circuits for each angle for any non-equivalent atom have been determined. The results of the topological analysis can be summarized as follows: (i) Despite the differences in space group symmetry Na2Ba6Si4O15 and β-Na2Ca6Si4O15 represent the same mixed-framework type, i.e. they are topologically equivalent. (ii) Although α- and β-Na2Ca6Si4O15 both are based on kröhnkite-type chains their mixed frameworks are significantly different, i.e. a potential α→β transition requires a larger re-arrangement of the structure and not only small shifts or tilts of the polyhedral units. (iii) The framework within the crystal structure of K2Ca6Si4O15 in turn is not topologically equivalent to those observed in the other three phases.
Furthermore, so-called polyhedral microensembles or PME’s were constructed for β-Na2Ca6Si4O15. On the lowest sublevel they are formed for each octahedron and tetrahedron in the asymmetric unit by considering all directly bonded [MO6]- and [TO4]-groups. They represent a geometrical interpretation of the coordination sequences up to the index k = 3. The PME’s of the first sublevel observed for the M nodes can be described as follows: each [MO6]-octahedron is immediately linked to six tetrahedra (see Fig. 4a). According to the classification given by Ilyushin and Blatov (2002) based on the calculation of the coordination sequences up to k = 3 (including the oxygen atoms) the PME’s can be denoted as {6,6,18} (for Ca(2) and Ca(4)). For the PME’s of the four crystallographically independent tetrahedral T (Si) nodes two groups of ensembles can be distigushed: {4,4,20} (for Si(1) and Si(2)) as well as {4,3,13} (for Si(3) and Si(4)) (see Fig. 4b and c).

PME’s for the M- and the T-nodes in β-Na2Ca6Si4O15 drawn in polyhedral representations. (a) {6,6,18} (for Ca(2) and Ca(4)), (b) {4,4,20} (for Si(1) and Si(2)) as well as (c) {4,3,13} (for Si(3) and Si(4))
The similarity between the values of the lattice parameters of Na2Ba6Si4O15 and β-Na2Ca6Si4O15 as well as the topological equivalence of their mixed frameworks prompted us to look for further structural correlations based on group theoretical considerations. First of all, the space group types of Na2Ba6Si4O15 (P121/c1) and β-Na2Ca6Si4O15 (P1c1) are in a group-subgroup relationship, i.e. P1c1 is a translationengleiche subgroup of index 2 of P121/c1. For a more detailed analysis the program STRUCTURE RELATIONS available via the web site of the Bilbao Crystallographic Server was employed (Tasci et al. 2012). The software is able to find relations between a crystal structure S with space group symmetry H when it can be derived from a virtual or real parent structure in a higher symmetry G using a left coset decomposition of G with respect to H: G = H + g2H +…+ gnH. In the next step the structure S to be tested is compared with the transformed structures giS. If the differences between the corresponding pairs of atoms in S and giS are below a certain threshold Δmax, a relationship is indicated. In the present case a relation between Na2Ba6Si4O15 and β-Na2Ca6Si4O15 is suggested if a maximum tolerance of 2 Å for the shifts between corresponding atoms is allowed. Both structures are related according to the following transformation (P,p): a,b,c (0.0041,-¼,-0.0098), i.e. the sequence of the lattice parameters is not changed. However, an origin shift has to be applied. The deviations in Ångstrom between the positions of the corresponding atoms for the different atom types in the Na2Ba6Si4O15 and β-Na2Ca6Si4O15 structures are as follows: tetrahedrally coordinated cations (spread: 0.158–0.568; average: 0.364), octahedrally coordinated cations (spread: 0.082; average: 0.082), non-framework cations (spread: 0.107 – 0.828; average: 0.356) and oxygen anions (spread: 0.134–1.697; average: 0.601). It is interesting to note, that the largest shift of about 1.7 Å occurs between the bridging oxygen atoms of the [Si2O7]–dimers. However, the average displacement of all corresponding atoms in both structures is relatively small and has a value of 0.172 Å.
Discussion
As mentioned in the experimental section a pronounced weight loss due to evaporation of Na2O was observed shifting the bulk composition of the sample away from Na2Ca3Si2O8 located on the tie-line Ca2SiO4 – Na2CaSiO4. This result is also of importance, because previous synthesis experiments by Maki (1969) and other groups were performed under similar conditions concerning temperature treatment of the samples. Therefore, it can be anticipated that the above mentioned substantial Na2O losses must have occurred in the older experiments as well. Na2Ca3Si2O8 (starting composition and chemical formula suggested by Maki) and Na2Ca6Si4O15 (the composition determined for our crystals) can be related by the following equation: 2 Na2Ca3Si2O8 -> Na2Ca6Si4O15 + Na2O, i.e. the requested change of the bulk composition to produce a phase pure Na2Ca6Si4O15 sample could be simply induced by partial volatilization of the sodium oxide component. Of course, in case that the Na2O losses are lower than needed for the reaction to be complete other phases such as Na2CaSiO4 or Ca2SiO4, for example, have to be present as well.
Furthermore, there is a striking similarity between Maki’s postulated monoclinic Na2Ca3Si2O8-phase and β-Na2Ca6Si4O15 not only what concerns the unit cell parameters but also what concerns the principle distribution of the relative intensities. The comparison between the calculated powder diffraction pattern of β-Na2Ca6Si4O15 and the data for Na2Ca3Si2O8 based on the PDF-entry 00-023-0670 is given in Fig. 5. The remaining small differences could be due to slightly different distributions of the Na/Ca atoms on the non-tetrahedrally coordinated sites, for example. In summary one can say that there are strong indications that monoclinic Na2Ca3Si2O8 is actually misinterpreted β-Na2Ca6Si4O15.

Comparison between (a) the simulated powder diffraction pattern of β-Na2Ca6Si4O15 based on the crystallographic data given in Tables 1 and 2; for the calculations the program FullProf.2 k (Rodriguez-Carvajal 2011) has been employed (λ = 1.54056 Å, Bragg-Brentano geometry, TCH-pseudo Voigt functions) and (b) the experimental powder pattern from PDF-entry no. 00-023-0670 for monoclinic Na2Ca3Si2O8 based on the data reported by Maki (1969)
This leaves the question whether Maki’s triclinic Na2Ca3Si2O8 polymorph may de facto also correspond to a further Na2Ca6Si4O15 modification. If so, several very recent publications on the luminescent properties of REE-doped Na2Ca3Si2O8 where phase identification was solely based on the evaluation of powder diffraction patterns using the PDF-entry 00-023-0668 have to be revised (Jin et al. 2015; Fu and Hu 2016).
Finally, we would like to point out that single-crystals of α-Na2Ca6Si4O15 were obtained by Armbruster and Röthlisberger (1990) as a by-product in experiments aiming on the synthesis of silicate-germanate melilite-type compounds using NaOH solutions under hydrothermal conditions (0.2 GPa, 700 °C). So far, however, this compound has not been mentioned in any of the previous phase equilibrium studies. The results of our investigation demonstrate that at least another polymorph of Na2Ca6Si4O15 can be prepared by standard ceramic methods without any application of pressure. This indicates that Na2Ca6Si4O15 has a stability field in the system Na2O-CaO-SiO2 at ambient pressure and that additional phase analytical studies are necessary to clarify this point. Notably, more than ninety years after the first comprehensive study of Morey and Bowen (1925) this ternary system still holds many surprises.
References
Armbruster T, Röthlisberger F (1990) Crystal growth and structures of mixed-anion silicates-germanates; Ca5[(Ge, Si)2O7][(Ge, Si)O4] and Na2Ca6[Si2O7][SiO4]2. Am Mineral 75:963–969
Arroyabe E, Kaindl R, Többens DM, Kahlenberg V (2009) K2Ca6Si4O15- structural and spectroscopical studies on a mixed tetrahedral-octahedral framework. J Solid State Chem 182:3254–3261
Blatov VA (2012) Nanocluster analysis of intermetallic structures with the program package TOPOS. Struct Chem 23:955–963
Burla MC, Caliandro R, Camalli M, Carrozzini B, Cascarano GL, De Caro L, Giacovazzo C, Polidori G, Spagna R (2005) SIR2004: an improved tool for crystal structure determination and refinement. J Appl Cryst 38:381–388
Dollase WA, Ross CR (1991) Crystal structure of orientationally disordered Na2(Ca, Sr)SiO4. Z Kristallogr 197:13–26
Dowty E (2011) ATOMS6.4. Shape Software, Kingsport
Farrugia LS (1999) WinGX suite for small-molecule single-crystal crystallography. J Appl Cryst 32:837–838
Fischer RX, Tillmanns E (1984) Die Kristallstruktur von Na4CaSi3O9 und dessen strukturelle Beziehungen zu K4SrGe3O9 und Ca3Al2O6. Z Kristallogr 166:245–256, in German
Fischer RX, Tillmanns E (1987) Revised data for combeite Na2Ca2Si3O9. Acta Cryst C 43:1852–1854
Fu Y, Hu Y (2016) A blue-green-emitting phosphor Na2Ca3Si2O8:Tb3+ with tunable emission color manipulated by cross-relaxation. J Mater Sci Mater Electron 27:3867–3872. doi:10.1007/s10854-015-4235-1
Garza Montoya B, Torres-Martinez LM, Quintana P, Ibarra J (2003) Alternative batch composition in the glass-forming region of the Na2O-CaO-SiO2 system. J Non-Cryst Solids 329:22–26
Ihara M, Odani K, Yoshida N, Kukunaga K, Setoguchi M, Higashi T (1984) The crystal structure of devitrite (disodium tricalcium hexasilicate), Ca3Na2Si6O16. J Ceram Assoc Japan 92:373–378
Ilyushin GD, Blatov VA (2002) Crystal chemistry of zirconosilicates and their analogs: topological classification of MT frameworks and suprapolyhedral invariants. Acta Cryst B 58:198–218
Jin Y, Fu Y, Hu Y, Chen L, Ju G, Mu Z (2015) Preparation and characterization of a long persistent phosphor Na2Ca3Si2O8:Ce3+. Opt Mater Express 5:1488–1497
Kahlenberg V, Hösch A (2002) The crystal structure of Na2Ca2Si2O7—a mixed anion silicate with defect perovskite characteristics. Z Kristallogr 217:155–163
Kahlenberg V, Girtler D, Arroyabe E, Kaindl R, Többens DM (2010) Devitrite (Na2Ca3Si6O16)-structural, spectroscopic and computational investigations on a crystalline impurity phase in industrial soda-lime glasses. Mineral Petrol 100:1–9
Knowles KM, Thompson RP (2014) Growth of devitrite, Na2Ca3Si6O16, in soda-lime-silica glass. J Am Ceram Soc 97:1425–1433
Kolitsch U, Fleck M (2006) Third update on compounds with kröhnkite-type chains: the crystal structure of wendwilsonite [Ca2Mg(AsO4)∙22H2O] and the new triclinic structure types of synthetic AgSc(CrO4)∙22H2O and M2Cu(Cr2O7)∙22H2O (M = Rb, Cs). Eur J Mineral 18:471–482
Kuo Y-M (2014) Role of sodium ions in the vitrification process: glass matrix modification, slag structure depolymerization, and influence of metal immobilization. J Air Waste Manage 64:774–784
Liebau F (1985) Structural chemistry of silicates. Springer Verlag, Berlin
Maki I (1969) Some 2CaO.SiO2 analogous compounds containing alkali oxide 2CaO.SiO2 systems. Semento Gijutsu Nempo 23:27–30, in Japanese
Moir GK, Glasser FP (1974) Phase equilibria in the system Na2SiO3-CaSiO3. Phys Chem Glasses 14:6–11
Morey GW, Bowen NL (1925) The ternary system sodium metasilicate - calcium metalicate -silica. J Soc Glass Tech 9:226–264
Ohsato H, Maki I, Takéuchi Y (1985) Structure of Na2CaSi2O6. Acta Cryst C 41:1575–1577
Ohsato H, Takéuchi Y, Maki I (1990) Structural study of the phase transition of Na4Ca4(Si6O18). Acta Cryst B 46:125–131
Robinson K, Gibbs GV, Ribbe PH (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science 172:567–570
Rodriguez-Carjaval J (2011) FullProf.2k, Version 5.00, Jan2011-ILL JRC. Institut Laue-Langevin, Grenoble, France
Segnit ER (1953) Further data on the system Na2O-CaO-SiO2. Am J Science 251:586–601
Shahid KA, Glasser FP (1970) The Na2O-CaO-5SiO2 phase in the system Na2O-CaO-SiO2. J Am Ceram Soc 53:423
Shahid KA, Glasser FP (1971) Phase equilibria in the glass forming region of the system Na2O-CaO-SiO2. Phys Chem Glasses 12:50–57
Shelby JE (2009) Introduction to glass science and technology, 2nd edn. The Royal Chemical Society, Cambridge
Sheldrick GM (1997) SHELX-97 - A program for crystal structure refinement. University of Göttingen, Germany.
Spek AL (2009) Structure validation in chemical crystallography. Acta Cryst D 65:148–155
Stoe & Cie GmbH (2005) X-Area Version 1.39, Darmstadt, Germany
Tamazyan RA, Malinovskii A, Sirota MJ (1987) Crystal structure and twinning of Na2Ba6(Si2O7)(Si O4)2. Sov Phys Crystallogr 32:519–522
Tasci ES, de la Flor G, Orobengoa D, Capillas C, Perez-Mato JM, Aroyo, MI (2012) An introduction to the tools hosted in the Bilbao Crystallographic Server. EPJ Web of Conferences, 22, 00009, 1–22
Thilo E (1951) Wege zu einer Konstitutionschemie der Silicate. Angew Chem 63:201–206 (in German)
Treushnikov EN, Ilyukhin VV, Belov NV (1971) Crystal structure of Na, Ca triorthosilicate, Na2Ca3(Si3O10). Sov Phys Crystallogr 16:56–58
Varshneya AK (1994) Fundamentals of inorganic glasses. Academic, San Diego
Wilson AJC (ed) (1995) International Tables for Crystallography, Volume C. Kluwer Academic Publishers, Dordrecht
Zhang Z, Xiao Y, Yang Y, Boom R, Voncken JHL (2009) Vitrified bottom ash slag from municipal solid waste incinerators - phase relations of CaO-SiO2-Na2O oxide system. VIII International Conference on Molten Slags, Fluxes and Salts – MOLTEN 2009, Santiago, Chile, 43–50.
Zhang Z, Xiao Y, Voncken J, Yang Y, Boom R, Wang N, Zou Z (2011) Phase equilibria in the Na2O-CaO-SiO2 system. J Am Ceram Soc 94:3088–3093
Acknowledgments
Open access funding provided by University of Innsbruck.The authors gratefully acknowledge the help of Dr. Takaya Nagai (Hokkaido University, Sapporo) for translating relevant paragraphs of Maki’s paper originally published in Japanese.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: M. A.T.M. Broekmans
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(TXT 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kahlenberg, V., Maier, M. On the existence of a high-temperature polymorph of Na2Ca6Si4O15—implications for the phase equilibria in the system Na2O-CaO-SiO2 . Miner Petrol 110, 905–915 (2016). https://doi.org/10.1007/s00710-016-0447-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00710-016-0447-1