
Abstract
Phosphonium phenolate zwitterions have been synthesized from 2,4-di-tert-butyl-6-(diphenylphosphino)phenol and five different oxirane derivatives. The reaction does not proceed at a detectable rate when the two reactants are combined in dichloromethane at room temperature. Despite the substantial ring strain, the reaction proceeds only with the addition of methanol, which acts as a hydrogen transfer shuttle, allowing a slow conversion to the desired zwitterions. The compounds have been fully characterized and single crystal X-ray crystallography has been performed on the methyloxirane and the phenyl glycidyl ether-derived zwitterion. The phosphonium phenolate units exhibit an ylidic bonding situation as evidenced by spectroscopic and crystallographic analysis. Glycidyl ethers were found to react faster than alkyl and aryloxiranes. Decomposition studies of the zwitterions showed high thermal stability in solution under ambient conditions. Under forced conditions (150 °C, 6 h), decomposition to the corresponding phosphine oxide and secondary aliphatic alcohols, the formally hydrogenated oxirane derivative, was observed.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epoxy molding compounds used to protect semiconductor circuits from environmental factors such as moisture, heat, and shock are typically based on diglycidyl ethers of various bisphenol A derivatives and so-called phenol-aralkyl resins, which are products of the condensation reaction between aldehydes and alkylphenols, as hardeners. In addition, additives and fillers are used, and an important component is the so-called accelerator, which accelerates the curing of the resin typically at temperatures above 150 °C and increases the number of molding cycles for mass production [1,2,3,4,5]. In particular, triarylphosphine accelerated formulations have optimal physical properties when cured, which subsequently improves the reliability of the encapsulated semiconductors [6,7,8,9] and recent work is attempting to further improve their properties [10,11,12]. Mechanistically, the accelerated curing of the epoxy-phenol reaction is explained by the initial formation of betaine I (Scheme 1) by nucleophilic attack of the phosphine from the side opposite to the oxygen of the oxirane group onto the sterically less hindered carbon atom of the oxirane ring. In turn, the Brønsted-basic zwitterion I undergoes an acid–base reaction with the phenols to form ion pair II. The resulting phenolates are more reactive with epoxy groups than phenols, which explains the faster curing [1,2,3,4].

Apparently, tertiary aromatic phosphines work as intended in these formulations, although it has long been known that epoxides and tertiary phosphines react to give the corresponding deoxygenated alkene and phosphine oxide as products [13,14,15]. The reaction mechanism has been studied theoretically [16] and catalyzed versions of the reaction have been disclosed [17,18,19]. The overall reaction is described by four steps and starts with the phosphine’s attack of the oxirane forming the same zwitterionic structure I as in the first step of the epoxy phenol reaction (Scheme 1). In a next step, the betaine I is transformed by rotation into the betaine Ia, which then forms the oxaphosphetane derivative III. Finally III decomposes to release the end-products, the corresponding phosphine oxide and the alkene.
Here we would like to report our contribution to a better understanding of the reaction of epoxides with phosphines [20]. We have chosen a (diphenylphosphino)phenol derivative combining the phenol and the phosphine into a single molecule [21] to study in particular the first step of the reaction and found evidence for the elusive formation of phosphonium phenolate species reminiscent of II. The resulting zwitterionic molecules do not undergo the deoxygenation reaction and are thermally quite stable, making them potentially interesting as catalysts for various reactions such as the synthesis of cyclic carbonates from epoxides and CO2 [22, 23] or organophotoredox catalysis [24, 25].
Results and discussion
First we tested triphenylphosphine (PPh3) and 2,4-di-tert-butyl-6-(diphenylphosphino)phenol (1) as initiators for the homopolymerization of phenyl glycidyl ether (PGE). 4-(Dimethylamino)pyridine (DMAP) was used as a benchmark and the activity of the initiators (5 mol% with respect to PGE) was investigated by dynamic differential scanning calorimetry (DSC) accompanied by on-line monitoring of sample weights in a simultaneous thermal analysis (STA) instrument. The results are shown in Fig. 1. In the DMAP case, an onset of the polymerization was observed at about 80 °C. The reaction showed its maximum heat flow at 129 ± 1 °C and a heat of polymerization of about 96 ± 3 kJ mol−1 was determined. At 210 °C a mass loss of 16 ± 2% was observed, which was attributed to the evaporation of PGE not yet incorporated into the macromolecules [26]. In the case of PPh3 no heat of polymerization could be observed, but only the evaporation of PGE as evidenced by the endothermic heat flow signal peaking at 193 °C and the pronounced mass loss resulting in about 7% residual mass at 230 °C.

Dynamic STA measurements of the homopolymerization of PGE initiated with DMAP, PPh3, and 1 (5 mol% in respect to PGE; heat rate = 10 K min.−1) showing the heat flow of DSC experiments (thick lines with symbols, exothermal reactions show a positive heat flow) and the masses of the samples in dependence of the temperature (dashed lines with the same color code)
With 1 as the initiator, diminished mass loss (about 13% residual mass at 230 °C) and a less pronounced endothermic heat flow with a peak at a higher temperature (209 °C) were observed than in the case of PPh3. Accordingly, both phosphines are not well suited to initiate the homopolymerization of PGE under these conditions.
In a next step, we investigated the reaction of PGE with 5 mol% of the phosphines at room temperature and 80 °C. In each case, no polymerization of PGE could be detected after 24 h and most of the PGE remained unreacted. For the reaction with PPh3 at room temperature, two thirds of the PPh3 was still present in the reaction mixture and one third was oxidized to triphenylphosphine oxide (OPPh3), as revealed by 31P NMR spectroscopy (see Fig. S2). Small amounts of (allyloxy)benzene were observed in the proton NMR spectrum ((allyloxy)benzene was identified by comparison to published NMR data [27]). At 80 °C PPh3 was fully converted to OPPh3 and (allyloxy)benzene was clearly detected in the proton NMR spectrum. Integration revealed an OPPh3: (allyloxy)benzene ratio of 2.5: 1, suggesting that OPPh3 was formed by direct oxidation and by the deoxygenation reaction responsible for (allyloxy)benzene formation. In contrast, no (allyloxy)benzene was detected with 1 as the phosphine. After 24 h at room temperature, 74% of 1 was still present and only 7% of the corresponding phosphine oxide was found [28]. In addition, a previously unknown signal was detected in the 31P NMR spectrum, which accounted for 19% of the phosphine signals. At 80 °C, about 80% of this unknown substance formed together with about 8% of the oxide of 1 and several other unknown phosphorus signals (12% in total; see Fig. S10).
As a next step, we sought to isolate and identify the unknown main product, which we hypothesized to be the corresponding phosphonium phenolate zwitterion resulting from the ring opening of PGE by 1 followed by hydrogen transfer. Similar zwitterionic species have been prepared by reaction of 1 with Michael acceptors [29], and we anticipated a similar reactivity with epoxides. In case of using epoxides, such reaction is additionally driven by the release of ring strain (about 113 kJ/mol) [30, 31]. However, when 1 was reacted with PGE at ambient conditions in dichloromethane (c = 1.3 mol/dm3) no conversion to the above detected species was observed over one week. Drawing parallels to the formation of zwitterions with Michael acceptors [29], the lack of reactivity is likely due to hindered hydrogen transfer from the phenol derivative 1 to the alkoxide in I (see Scheme 1). Accordingly, 5 equivalents of methanol were added to the reaction mixture and slow formation of the desired product was observed at room temperature. Besides PGE, methyloxirane, ethyloxirane, phenyloxirane, and (butoxymethyl)oxirane were also used as substrates and in all cases the addition of methanol was necessary to achieve conversion to the desired products (Scheme 2). In general, the reaction rate is low and a quantitative conversion of 1 cannot be achieved within a week at room temperature. No by-product formation has been observed under these conditions. Therefore, a typical preparation of the zwitterions is carried out at room temperature using the lowest possible dilution (solvent: a mixture of dichloromethane and 5 equiv. of methanol in respect to 1) for 7 days. The remaining starting materials are then removed by recrystallization, resulting in moderate (non optimized) yields of the zwitterions 2a-2e (see Table 1).

The compounds were characterized by 1H, 13C, and 31P NMR spectroscopy and the final proof of the molecular structure was obtained by single crystal X-ray structure determination (vide infra). In the 1H NMR spectrum, a characteristic doublet of doublets signal is observed in the range of 6.10 to 6.15 ppm for the zwitterions 2a-2e (see Table 1). This resonance is attributed to the proton at position 5 in the 2,4-di-tert-butylphenolate substituent of the zwitterions and involves meta-coupling to the proton at position 3 (4JHH ≈ 2.5 Hz) as well as coupling to phosphorus (3JPH ≈ 14.5 Hz). For comparison, the same proton in the parent phosphine 1 shows a resonance at 6.88 ppm [32]. In the Michael acceptor-derived zwitterions, this proton is similarly shifted upfield as in 2a-2e and is found in the range of 6.09 to 6.21 ppm [29].
The 13C NMR spectrum of 2d is shown in Fig. 2 and is representative of all zwitterions disclosed herein. The carbon atom in position 1 of the 2,4-di-tert-butylphenolate substituent shows a doublet (2JPC ≈ 4.5 Hz) in the range of 172.8 to 173.7 ppm (Table 1), which is indicative of a quinonic resonance structure, since benzoquinones show shifts of about 188 ppm, whereas hydroquinones appear at about 150 ppm [33] (in the parent phosphine 1 the corresponding peak is observed at 155.9 ppm [32]). The most characteristic chemical shift for the zwitterions 2a-2e in the 13C NMR spectra is found for the resonances of carbon 6, which are observed in the range of 100.4 to 100.9 ppm (1JPC ≈ 95 Hz). As expected, the diastereotopic phenyl substituents give rise to two distinct sets of 13C signals as shown in Fig. 2. The 13C chemical shifts for the methylene carbon 15 (directly attached to the phosphorus atom) are found in the range of 34.7 to 40.7 ppm (1JPC between 64 and 54 Hz). For comparison, the corresponding carbon atom in similar alkyltriphenylphosphonium halides resonates at 18 to 27 ppm (1JPC ≈ 54 Hz) [34, 35].

13C{1H} NMR spectrum of 2d in CDCl3 recorded at 100 MHz; numbering in accordance with the numbering of the atoms in the molecular structures retrieved from single crystal X-ray measurements
The 31P NMR shift (against H3PO4, 85%) of the adducts is in the range of 18.3 to 18.9 ppm, which is slightly shifted upfield compared to the corresponding values for the Michael acceptor derived zwitterions of 1 [29]. For comparison, phosphine 1 has a 31P NMR shift of −29.7 ppm [32].
Crystals suitable for single crystal X-ray diffraction analysis were obtained from compounds 2a and 2d. Crystals of 2a were grown from a concentrated solution in toluene/THF (3:1) and two crystallographically independent conformers of 2a were found in the unit cell. As shown in Fig. 3, one conformer exhibits an intramolecular hydrogen bonding interaction between the phenolate oxygen (O1) and the proton of the hydroxyl group (attached to O2) resulting in an O1-O2 distance of 2.583(5) Å. The second conformer crystallizes in a hydrogen bonded coordination polymer in which the phenolate oxygen (O3) is 2.596(5) Å apart from the oxygen atom (O4) of the hydroxyl group of the next repeating unit. In 2d, the conformer with the intramolecular hydrogen bond was found with a O1-O2 distance of 2.519(4) Å (Fig. 2, Gy box). The P1–O1 distances of 2.813(4) Å (2a, intramolecular hydrogen bonding) and 2.708(4) Å (2a, intermolecular hydrogen bonding) as well as 2.826(3) Å in 2d suggest a weak binding interaction between the phenolate and the phosphonium center as observed in similar zwitterions with P–O distances in the range of 2.60–2.95 Å [26, 36, 37] (Fig. 3, ochre box). The P1-O2 distance is with 3.500(3) Å (in 2d) or 3.313(4) Å and 3.574(4) Å (2a chain conformer and 2a ring conformer) significantly higher. In any case, the P-O distances are significantly larger than expected for covalent P-O bonds in 1,2-oxaphosphetanes [38].

Results of single crystal X-ray diffraction measurements showing the molecular structures of the two independent conformers of 2a present in the crystal; above: intramolecular hydrogen bonding forms a 8-membered ring; below: intermolecular hydrogen bonding results in a coordination polymer and of 2d (shown in the gray box); the sketch in ochre box illustrates the bonding situation of the phosphorous atom
This weak interaction of P1 and O1 causes a slight distortion of the tetrahedral bonding geometry around the phosphonium atom toward a distorted trigonal pyramidal geometry with C24 and O1 as the apexes, which can be seen by the fact that each of the atoms C6, C15 and C18 have angles smaller than 109.5° with the atom C24 (e.g. C6-P1-C24 = 107.3(3)°, C15-P1-C24 104.3(3)°, C18-P1-C24 106.2(3)° in 2a with intramolecular hydrogen bonding). Furthermore, the P1-C24 distance is always slightly larger than the distance between P1 and C18 (e.g. 1.825(5) Å and 1.813(5) Å in 2a with intramolecular hydrogen bonding). Finally, the O1-P1-C18 angles are between 163° and 165°, and the torsion angle defined by O1-C1-C6-P1 is in all cases less than 7°.
The bonding situation in the phenolate ring is similar to what have been found for Michael-acceptor derived zwitterions [29] or for (triphenylphosphonium)phenolate and point to an electron delocalization within a ylidic system [25, 36]. The P1-C6 distances in 2a (1.774(4) Å for the conformer with intramolecular hydrogen bonding and 1.780(4) Å for the intermolecular hydrogen bonded molecules) and in 2d (1.778(4) Å) are significantly shorter than the corresponding P-C18 and P-C24 distances and shorter than in the free phosphine 1 (1.825 Å) [32]. The same accounts for the O1-C1 bond lengths, which are notably shorter in the zwitterions 2a (1.286(5) Å and 1.292(4) Å) and 2d (1.297(4) Å) than in the parent phosphine 1 (1.373(2) Å) [32].
All presented zwitterions exhibit similar UV–Vis spectral properties with a single absorption feature ranging from 310 to 410 nm, peaking at 346 ± 1 nm. The molar extinction coefficients are in between 5300 and 5800 dm3 mol−1 cm−1. Absorption spectra can be found in the supporting information (Fig. S47).
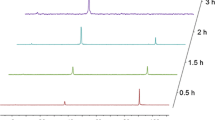
In order to estimate the relative reactivity of the epoxides studied, the formation of the zwitterions 2a-2e was followed over time. In contrast to the preparation of the zwitterions, we changed the amounts of reactants and used 10 equivalents of the respective epoxide and 10 equivalents of methanol (with respect to 1). Furthermore, a defined volume of mesitylene was added as an internal standard for integrating the 1H NMR spectra used to quantify the conversion after specific time intervals of the reaction carried out at room temperature. The zwitterions were formed exclusively, as no other products were observed in either the 1H NMR or 31P NMR spectra. The results are shown in Fig. 4 and show a faster reaction of glycidyl ethers than for methyloxirane, ethyloxirane and phenyloxirane. A similar reactivity trend was observed for epoxy-amine reactions [39].

Conversion of 1 into the corresponding zwitterions 2a-2e as determined by 1H NMR spectroscopy of the reaction mixture; reaction conditions: 1 equiv. 1, 10 equiv. oxirane derivative, 10 equiv. methanol, room temperature
Compound 2d was subjected to decomposition studies in three different NMR solvents, namely benzene-d6, CDCl3 and CD3CN. Solutions of 2d were either stored at room temperature or heated to 60 °C and the samples were subjected to 1H and 31P NMR analysis after specified time frames. At room temperature, no degradation was observed in any of the solvents after 7 days.
The same is true for solutions 2d in CDCl3 and CD3CN stored at 60 °C for 3 d. After 7 days at 60 °C, traces of phosphine oxide were observed in the CDCl3 solution, while in acetonitrile no decomposition was detected even after this time. It appears that epoxy-based phosphonium phenolate zwitterions are more stable than their Michael acceptor-derived counterparts, which already show decomposition/oxidation under similar conditions [29]. However, the benzene-d6 solution shows signs of decomposition earlier. After 7 d at 60 °C, some oxide of 1 (less than 3%) is visible in the 31P NMR spectrum and new peaks (multiplets at 3.44 and 3.85 ppm) are present in the 1H NMR spectrum. The characteristic 1H NMR signals of (allyloxy)benzene were not observed. In an attempt to identify the newly formed species, 2d was dissolved in CDCl3 and heated to 150 °C for 6 h in an autoclave. The resulting solution was directly subjected to NMR and GC–MS analysis and the main decomposition products could be identified as phenoxypropan-2-ol and the phosphine oxide formed from 1 [40] (Scheme 3).

Further decomposition products included phenol as well as other unidentified products in small amounts (in sum less than 15%, see Figs. S43 and S44). The formation of phenoxypropan-2-ol from 2d requires a formal reduction of the 2-hydroxy-3-phenoxypropyl group and oxidation of the phosphonium to a phosphine oxide. This redox reaction may be facilitated by water, as it has long been known that tertiary phosphines and water form the corresponding phosphine oxides and hydrogen under alkaline conditions [41]. Furthermore, it has been shown that the attack of water on phosphonium centers leads to the elimination of the corresponding newly formed phosphine oxide [42]. In a recent publication, this reactivity is exploited in a photocatalytic phosphine-mediated water activation for hydrogenation of alkenes [43].
Conclusion
The reaction of 2,4-di-tert-butyl-6-(diphenylphosphino)phenol with various oxirane derivatives yields phosphonium phenolate zwitterions. The use of a hydrogen transfer agent, in this case methanol, is necessary to facilitate the reaction at room temperature. Therefore, it can be said that the failure of PPh3 and 1 to promote the phenyl glycidyl ether homopolymerization is not due to the same degradation reaction. While PPh3 deactivates via Wittig chemistry, forming (allyloxy)benzene via the intermediate initial zwitterion, this degradation pathway is blocked by the addition of phenols. The acidic phenols protonate the alkoxide group present in the initially formed zwitterion, thereby inhibiting the oxaphosphetane formation ultimatively leading to the formation of olefins and phosphine oxides. The resulting phenolate is a much weaker nucleophile than the initially formed alkoxide, which explains the high temperatures required to cure epoxy-phenol formulations compared to the DMAP initiated homopolymerization. Phosphonium phenolates, as shown here for the zwitterion 2d, decompose via a different pathway resulting in the formation of the phosphine oxide and secondary alcohols resulting from the formal hydrogenation of the oxirane derivative.
Experimental
All experiments were performed under ambient conditions. Chemicals were purchased from Sigma-Aldrich, Carl Roth, Merck, or TCI and were used as received. 2,4-Di-tert-butyl-6-(diphenylphosphino)phenol (1) was prepared according to a published procedure [44]. NMR spectra were recorded on a Bruker AVANCE III 300 spectrometer or a JEOL JNM-ECZ 400 MHz spectrometer and are referenced to tetramethylsilane (1H, 13C), and 85% H3PO4 (31P). Deuterated solvents were obtained from Cambridge Isotope Laboratories Inc. UV–Vis spectra were recorded on an Agilent Cary 60 UV–Vis spectrophotometer. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) was performed with a Netzsch simultaneous thermal analyzer STA 449C (crucibles: aluminum from Netzsch). The heating rate was 10 °C/min until a final temperature of 550 °C was reached. A helium flow of 20 cm3 min−1 was used in combination with a protective flow of helium of 10 cm3 min−1. Typically, 200 mg (1 eq, 1.33 mmol) of PGE were mixed with the respective initiator (0.05 eq, 0.067 mmol) until a homogeneous mixture was obtained. From this solution, 3 to 15 mg were transferred into the aluminum crucible, which was then closed with the aid of a mechanical press. A hole was cut through the lid to enable evaporation. The crucible was then subjected to the temperature program specified above. For decomposition studies, 20 mg (0.037 mmol) of 2d were dissolved in 0.7 cm3 of the respective deuterated solvent (CDCl3, C6D6, or CD3CN). The solution was kept in an NMR tube either at room temperature or at 60 °C and subjected to 1H and 31P NMR analysis after specific time frames. The decomposition study using 2d at forcing conditions was carried out as follows: 30 mg 2d (0.06 mmol) were dissolved in 0.7 cm3 CDCl3 and heated to 150 °C for 6 h using a Monowave 50 reactor (Anton Paar). The resulting yellow solution was then directly subjected to NMR and GC–MS analysis using an Agilent Technologies 7890A GC system equipped with an Agilent Technologies J&W HP 5MS capillary column (30 m × 0.25 mm × 0.25 µm film with 5% phenyl- and 95% methylpolysiloxane). He 5.0 (Air Liquide) was used as carrier gas at a constant flow rate. Samples were injected in split mode using an Agilent Technologies 7683 Series autosampler and an Agilent Technologies 7683B Series injector. Methanol and ethyl acetate were used to flush the needle before and after injection. Masses were analyzed with a 5975C mass selective detector (inert MSD with Triple Axis Detector system) by electron-impact ionization with a potential of E = 70 eV. The following temperature program was used for all GC–MS measurements: 50 °C 1 min, followed by a linear heating ramp (40 °C per min until 300 °C) and 5 min at 300 °C, solvent delay: 4.0 min. For X-ray structure analyses the respective crystals were mounted onto the tips of glass fibres. Data collection was performed with a Bruker-AXS SMART APEX CCD diffractometer using graphite-monochromated Mo-Kα radiation (0.71073 Å). The data were reduced to Fo2 and corrected for absorption effects with SAINT (version 6.45, Bruker AXS Inc., 1997–2003) and SADABS (version 2.10. Bruker AXS Inc.), respectively [45]. The structures were solved by direct methods and refined by full-matrix least-squares method (SHELXL97 or SHELXL19) [46]. If not noted otherwise all non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were located in calculated positions to correspond to standard bond lengths and angles. Figures of solid state molecular structures were generated using VESTA [47]. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2330232 (2a) and 2330231 (2d).
2,4-Di- tert -butyl-6-[(2-hydroxypropyl)diphenylphosphonio]phenolate (2a, C 29 H 37 O 2 P)
Phosphine 1 (200 mg, 0.51 mmol, 1 eq) was dissolved in 2-methyloxirane (296 mg, 5.1 mmol, 10 eq) and methanol (82 mg, 2.6 mmol, 5 eq) in a 4 cm3 screw-cap vial. The reaction mixture was stirred at room temperature for 5 days. Afterwards all volatiles were evaporated in vacuo and the residue recrystallized by vapor diffusion of pentane on a concentrated solution in THF. The crystals were filtered off, washed with pentane and dried in vacuum. Yield: 147 mg (pale yellow crystals, 64.2%); 1H NMR (300 MHz, CDCl3, 298 K): δ = 1.05 (s, 9H, CCH3), 1.36–1.49 (m, 12H, CCH3 + C(OH)-CH3), 2.90–3.05 (m, 1H, CH2), 3.07–3.24 (m, 1H, CH2), 4.26–4.42 (m, 1H, (OH)CH), 6.10 (dd, 3JP-H = 14.8 Hz, 4JH-H = 2.4 Hz, 1H, H5), 7.34–7.44 (m, 2H, Ar–H), 7.46 (d, 1H, 4JH-H = 2.4 Hz, H3), 7.48–7.57 (m, 6H, Ar–H), 7.58–7.67 (m, 2H, Ar–H) ppm; 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ = 25.2 (d, 3JP-C = 17.3 Hz, (OH)CCH3), 29.4 (s, CCH3), 31.4 (s, CCH3), 34.1 (d, 4JP-C = 1.2 Hz, CCH3), 35.3 (d, 4JP-C = 2.1 Hz, CCH3), 39.2 (d, 1JP-C = 63.9 Hz, PCH2), 61.4 (d, 2JP-C = 4.7 Hz, (OH)C), 100.9 (d, 1JP-C = 96.5 Hz, C6), 122.8 (d, 1JP-C = 88.3 Hz, Ci-Ph), 126.4 (d, 1JP-C = 85.4 Hz, Ci-Ph), 127.5 (d, 2JP-C = 12.6 Hz, C5), 129.3 (d, 3JP-C = 11.0 Hz, Cm-Ph), 129.5(d, 3JP-C = 11.0 Hz, Cm-Ph), 131.1 (d, 4JP-C = 1.6 Hz, C3), 132.6 (d, 2JP-C = 8.6 Hz, Co-Ph), 132.8 (d, 3JP-C = 2.8 Hz, Cm-Ph), 133.0 (d, 2JP-C = 9.6 Hz, Co-Ph), 134.9 (d, 3JP-C = 15.0 Hz, C4), 141.1 (d, 4JP-C = 7.8 Hz, C2), 172.8 (d, 2JP-C = 4.2 Hz, C1) ppm; 31P{1H} NMR (162 MHz, CDCl3, 298 K): δ = 18.3 ppm; UV–Vis (CHCl3): λmax = 346 nm (ε = 5.55 × 103 dm3 mol−1 cm−1).
2,4-Di- tert -butyl-6-[(2-hydroxybutyl)diphenylphosphonio]phenolate (2b, C 30 H 39 O 2 P)
Phosphine 1 (200 mg, 0.51 mmol, 1 eq) was dissolved in dichloromethane (0.3 cm3) in a 4 cm3 screw-cap vial and methanol (82 mg, 2.6 mmol, 5 eq.) was added. In a separate vial, 2-ethyloxirane (37 mg, 0.51 mmol, 1 eq.) was dissolved in dichloromethane (0.1 cm3) and the solution was added dropwise to the first solution. The reaction mixture was stirred at room temperature for 7 days. Afterwards all volatiles were evaporated in vacuo and the residue recrystallized via vapor diffusion of pentane on a concentrated solution in THF. The crystals were filtered off, washed with pentane and dried in vacuum. Yield: 85 mg (colorless microcrystals, 36.0%); 1H NMR (300 MHz, CDCl3, 298 K): δ = 0.99 (t, 3H, CH2CH3), 1.03 (s, 9H, CCH3), 1.41 (s, 9H, CCH3), 1.55–1.66 (m, 1H, (OH)CCH2), 1.68–1.80 (m, 1H, (OH)CCH2), 2.96–3.04 (m, 2H, PCH2), 4.01–4.14 (m, 1H, (OH)CH), 6.04 (dd, 3JP-H = 14.7 Hz, 4JH-H = 2.5 Hz, 1H, H5), 7.34–7.41 (m, 2H, Ar–H), 7.42 (d, 1H, 4JH-H = 2.7 Hz, H3), 7.47–7.54 (m, 6H, Ar–H), 7.57–7.64 (m, 2H, Ar–H), 8.06 (br, 1H, OH) ppm; 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ = 10.3 (s, CH2CH3), 29.4 (s, CCH3), 31.4 (s, CCH3), 31.8 (d, 3JP-C = 16.4 Hz, (OH)CCH2), 34.0 (d, 4JP-C = 1.5 Hz, CCH3), 35.2 (d, 4JP-C = 2.4 Hz, CCH3), 37.3 (d, 1JP-C = 57.8 Hz, PCH2), 66.1 (d, 2JP-C = 3.8 Hz, (OH)C), 100.3 (d, 1JP-C = 96.8 Hz, C6), 122.8 (d, 1JP-C = 88.6 Hz, Ci-Ph), 126.9 (d, 1JP-C = 84.8 Hz, Ci-Ph), 127.5 (d, 2JP-C = 13.0 Hz, C5), 129.3 (d, 3JP-C = 10.6 Hz, Cm-Ph), 129.4 (d, 3JP-C = 8.6 Hz, Cm-Ph), 131.0 (d, 4JP-C = 2.4 Hz, C3), 132.6 (d, 2JP-C = 8.7 Hz, Co-Ph), 132.7 (s, Cp-Ph), 132.8 (d, 3JP-C = 2.9 Hz, Cp-Ph), 133.0 (d, 2JP-C = 9.6 Hz, Co-Ph), 134.1 (d, 3JP-C = 14.9 Hz, C4), 140.9 (d, 3JP-C = 8.2 Hz, C2), 173.7 (d, 2JP-C = 4.8 Hz, C1) ppm; 31P{1H} NMR (162 MHz, CDCl3, 298 K): δ = 18.8 ppm; UV–Vis (CHCl3): λmax = 345 nm (ε = 5.69 × 103 dm3 mol−1 cm−1).
2,4-Di- tert -butyl-6-[(2-hydroxy-2-phenylethyl)diphenylphosphonio]phenolate (2c, C 34 H 39 O 2 P)
Synthesis was carried out in the same way as described for 2b, using styrene oxide (61 mg, 0.51 mmol, 1 eq.). The residue obtained after evaporation of volatiles was recrystallized from a hot toluene/THF mixture. Yield: 124 mg (orange solid, 47.6%); 1H NMR (300 MHz, CDCl3, 298 K): δ = 1.11 (s, 9H, CCH3), 1.51 (s, 9H, CCH3), 3.15–3.29 (m, 1H, CH2), 3.34–3.52 (m, 1H, CH2), 5.29 (dd, 1H, 3JH-H = 12.4, 9.3 Hz, CH), 6.15 (dd, 3JP-H = 14.7 Hz, 4JH-H = 2.3 Hz, 1H, H5), 7.28–7.43 (m, 5H, Ar–H), 7.45–7.57 (m, 5H, Ar–H), 7.58–7.76 (m, 6H, Ar–H), 8.80 (br, 1H, OH) ppm; 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ = 29.3 (s, CCH3), 31.4 (s, CCH3), 34.0 (d, 4JP-C = 1.1 Hz, CCH3), 35.2 (d, 4JP-C = 2.1 Hz, CCH3), 40.7 (d, 1JP-C = 53.8 Hz, PCH2), 67.7 (d, 2JP-C = 3.8 Hz, (OH)C), 100.4 (d, 1JP-C = 96.5 Hz, C6), 122.4 (d, 1JP-C = 88.9 Hz, Ci-Ph), 125.8 (s, Ar–C), 126.5 (d, 1JP-C = 84.5 Hz, Ci-Ph), 127.4 (s, C5), 127.6 (s, Ar–C), 128.7 (s, Ar–C), 129.2 (d, 3JP-C = 11.3 Hz, Cm-Ph), 129.5 (d, 3JP-C = 12.4 Hz, Cm-Ph), 131.2 (d, 4JP-C = 1.9 Hz, C3), 132.5 (d, 2JP-C = 8.5 Hz, Co-Ph), 132.7 (d, 4JP-C = 2.8 Hz, Cp-Ph), 133.0 (d, 4JP-C = 3.0 Hz, Cp-Ph), 133.0 (d, 2JP-C = 9.6 Hz, Co-Ph), 134.5 (d, 3JP-C = 15.1 Hz, C4), 141.0 (d, 3JP-C = 7.8 Hz, C2), 145.1 (d, 3JP-C = 15.7 Hz, (OH)CHCAr), 173.6 (d, 2JP-C = 5.0 Hz, C1) ppm; 31P{1H} NMR (162 MHz, CDCl3, 298 K): δ = 18.3 ppm; UV–Vis (CHCl3): λmax = 345 nm (ε = 5.34 × 103 dm3 mol−1 cm−1).
2,4-Di- tert -butyl-6-[(2-hydroxy-3-phenoxypropyl)diphenylphosphonio]phenolate (2d, C 35 H 41 O 3 P)
Synthesis was carried out in the same way as described for 2b, using phenyl glycidyl ether (77 mg, 0.51 mmol, 1 eq). The obtained residue after evaporation of volatiles was recrystallized from hot toluene. Yield: 162 mg (pale yellow crystals, 58.7%); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.05 (s, 9H, CCH3), 1.44 (s, 9H, CCH3), 2.95–9.10 (m, 1H, P-CH2), 3.49–3.61 (m, 1H, P-CH2), 3.84–3.94 (m, 1H, O-CH2), 4.27–4.36 (m, 1H, O-CH2), 4.42–4.56 (m, 1H, (OH)CH), 6.07 (dd, 3JP-H = 14.9 Hz, 4JH-H = 2.5 Hz, 1H, H5), 6.87–6.99 (m, 3H, Ar–H), 7.23–7.31 (m, 2H, Ar–H), 7.36–7.65 (m, 11H, Ar–H) ppm; 13C{1H} NMR (100 MHz, CDCl3, 298 K): δ = 29.3 (s, CCH3), 31.4 (s, CCH3), 34.0 (d, 4JP-C = 1.5 Hz, CCH3), 34.8 (d, 1JP-C = 60.2 Hz, PCH2), 35.3 (d, 4JP-C = 2.4 Hz, CCH3), 63.9 (d, 2JP-C = 3.9 Hz, (OH)C), 70.9 (d, 3JP-C = 15.9 Hz, COPh), 100.7 (d, 1JP-C = 97.3 Hz, C6), 114.5 (s, OPhortho), 121.2 (s, OPhpara), 122.5 (d, 1JP-C = 88.1 Hz, Ci-Ph), 126.4 (d, 1JP-C = 86.2 Hz, Ci-Ph), 127.5 (d, 2JP-C = 12.5 Hz, C5), 129.3 (d, 3JP-C = 2.9 Hz, Cm-Ph), 129.4 (d, 2JP-C = 3.9 Hz, Cm-Ph), 129.7 (s, OPhmeta), 131.2 (d, 4JP-C = 2.4 Hz, C3), 132.6 (d, 2JP-C = 9.2 Hz, Co-Ph), 132.8 (d, 4JP-C = 2.9 Hz, Cp-Ph), 132.9 (d, 4JP-C = 2.9 Hz, Cp-Ph), 133.1 (d, 2JP-C = 9.2 Hz, Co-Ph), 134.6 (d, 3JP-C = 14.9 Hz, C4), 141.0 (d, 3JP-C = 7.7 Hz, C2), 158.2 (s, OPhipso), 173.4 (d, 2JP-C = 4.8 Hz, C1) ppm; 31P{1H} NMR (162 MHz, CDCl3, 298 K): δ = 18.7 ppm; UV–Vis (CHCl3): λmax = 347 nm (ε = 5.75 × 103 dm3 mol−1 cm−1).
2-[(3-Butoxy-2-hydroxypropyl)diphenylphosphonio]-4,6-di- tert -butylphenolate (2e, C 33 H 45 O 3 P) 2e
was prepared analogously to 2b, using butyl glycidyl ether (66 mg, 0.51 mmol, 1 eq.). The residue obtained after evaporation of volatiles was recrystallized by vapor diffusion of pentane on a concentrated solution in toluene. Yield: 89 mg (colorless solid, 33.5%); 1H NMR (400 MHz, CDCl3, 298 K): δ = 0.91 (t, 3JH-H = 7.3 Hz, 3H, CH2CH3), 1.03 (s, 9H, CCH3), 1.33–1.40 (m, 2H, CH2CH3), 1.41 (s, 9H, CCH3), 1.52–1.60 (m, 2H, CH2CH2), 2.85–2.98 (m, 1H, PCH2), 3.38 (m, 1H, (OH)CCH2), 3.43–3.49 (m, 3H, PCH2’ + OCH2), 3.62–3.69 (m, 1H, (OH)CCH2), 4.17–4.29 (m, 1H, (OH)CH), 6.04 (dd, 3JP-H = 14.7 Hz, 4JH-H = 2.7 Hz, 1H, H5), 7.37–7.45 (m, 3H, Ar–H), 7.46–7.52 (m, 4H, Ar–H), 7.53–7.64 (m, 4H, Ar–H), 8.23 (br, 1H, OH) ppm; 13C{1H} NMR (100 MHz, CDCl3, 298 K): δ = 14.1 (s, CH2CH3), 19.5 (s, CH2CH3), 29.4 (s, CCH3), 31.4 (s, CCH3), 32.0 (s, CH2CH2), 34.0 (d, 4JP-C = 1.2 Hz, CCH3), 34.7 (d, 1JP-C = 59.7 Hz, PCH2), 35.2 (d, 4JP-C = 2.4 Hz, CCH3), 64.6 (d, 2JP-C = 4.3 Hz, (OH)CH), 71.3 (s, OCH2), 74.5 (d, 3JP-C = 15.9 Hz, (OH)CHCO), 100.7 (d, 1JP-C = 97.8 Hz, C6), 123.3 (d, 1JP-C = 87.2 Hz, Ci-Ph), 126.7 (d, 1JP-C = 86.7 Hz, Ci-Ph), 127.5 (d, 2JP-C = 13.0 Hz, C5), 129.3 (d, 3JP-C = 11.6 Hz, Cm-Ph), 131.1 (d, 4JP-C = 2.4 Hz, C3), 132.6 (d, 2JP-C = 8.7 Hz, Co-Ph), 132.8 (d, 4JP-C = 2.9 Hz, Cp-Ph), 133.2 (d, 2JP-C = 9.2 Hz, Co-Ph), 134.3 (d, 2JP-C = 14.9 Hz, C4), 140.9 (d, 4JP-C = 7.7 Hz, C2), 173.6 (d, 2JP-C = 4.3 Hz, C1) ppm; 31P{1H} NMR (162 MHz, CDCl3, 298 K): δ = 18.9 ppm; UV–Vis (CHCl3): λmax = 347 nm (ε = 5.77 × 103 dm3 mol−1 cm−1).
Data availability
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
Kinjo N, Ogata M, Nishi K, Kaneda A, Dušek K (1989) Adv Polym Sci 88:1
Pham HQ, Marks MJ (2005) Epoxy Resins. In: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH
Karak N (2021) ACS Symp Ser 1385:1
Kim WG, Lee JY, Part KY (1993) J Polym Sci Part A: Polym Chem 31:633
Verma C, Olasunkanmi LO, Akpan ED, Quraishi MA, Dagdag O, El-Gouri M, Sherif ESM, Ebenso EE (2020) React Funct Polym 156:104741
Ryu JH, Choi KS, Kim WG (2005) J Appl Polym Sci 96:2287
Nagai A, Kokaku H, Ishii T (2002) J Appl Polym Sci 85:2335
Ogata M, Kinjo N, Eguchi S, Hozoji H, Kawata T, Sashima H (1992) J Appl Polym Sci 44:1795
Han S, Yoon HG, Suh KS, Kim WG, Moon TJ (2000) J Polym Sci Part A: Polym Chem 37:713
Su C-C, Wei C-H, Yang C-C (2013) Ind Eng Chem Res 52:2528
Tyberg CS, Shih P, Verghese N, Loos AC, Lesko J, Riffle JS (2000) Polymer 41:9033
Li J, Liu Y, Sun Z, Zhang M, Hong H, Moon KS, Wong C (2022) Chem Mater 34:3280
Boskin MJ, Denney DB (1959) Chem Ind 330
Speziale AJ, Bissing DE (1963) J Am Chem Soc 85:1888
Vedejs E, Fuchs PL (1973) J Am Chem Soc 95:822
Kalaiselvan A, Venuvanalingam P (2005) Tetrahedron Lett 46:4087
Zhu Z, Espenson JH (1995) J Mol Catal A 103:87
Gable KP, Brown EC (2003) Synlett 2243
Mitsudome T, Noujima A, Mikami Y, Mizugaki T, Jitsukawa K, Kaneda K (2010) Angew Chem Int Ed 49:5545
Romanchick WA, Sohn JE, Geibel JF (1983) ACS Symp Series 221:85
Heinicke JW (2024) Chem Eur J 30:e202302740
Büttner H, Steinbauer J, Wulf C, Dindaroglu M, Schmalz H-G, Werner T (2017) Chemsuschem 10:1076
Toda Y, Hashimoto K, Mori Y, Suga H (2020) J Org Chem 85:10980
Toda Y, Tanaka K, Matsuda R, Sakamoto T, Katsumi S, Shimizu M, Ito F, Suga H (2021) Chem Commun 57:3591
Toda Y, Kobayashi T, Hirai F, Yano T, Oikawa M, Sukegawa K, Shimizu M, Ito F, Suga H (2023) J Org Chem 88:9574
Edinger D, Fischer SM, Slugovc C (2024) Macromol Chem Phys 225:2300299
Noda H, Motokura K, Miyaji A, Baba T (2013) Adv Synth Catal 355:973
He L-P, Mu H-L, Li B-X, Li Y-S (2010) J Polym Sci Part A: Polym Chem 48:311
Steiner MR, Schmallegger M, Donner L, Hlina JA, Marschner C, Baumgartner J, Slugovc C (2024) Beilstein J Org Chem 20:41
Cremer D, Kraka E (1985) J Am Chem Soc 107:3800
Morgan KM, Ellis JA, Lee J, Fulton A, Wilson SL, Dupart PS, Dastoori R (2013) J Org Chem 78:4303
Heinicke J, Kadyrov R, Kindermann MK, Koesling M, Jones PG (1996) Chem Ber 129:1547
Kim B, Storch G, Banerjee G, Mercado BQ, Castillo-Lora J, Brudvig GW, Mayer JM, Miller SJ (2017) J Am Chem Soc 139:15239
Haitham E, Yaccoubi F (2023) Phosphorus, Sulfur Silicon Relat Elem 198:354
Mannu A, Di Pietro ME, Priola E, Baldino S, Sacchetti A, Mele A (2021) Res Chem Intermed 47:1663
Toda Y, Sakamoto T, Komiyama Y, Kikuchi A, Suga H (2017) ACS Catal 7:6150
Zhu X-F, Henry CE, Kwon OJ (2007) J Am Chem Soc 129:6722
Hamaguchi M, Iyama Y, Mochizuki E, Oshima T (2005) Tetrahedron Lett 46:8949
Ehlers J-E, Rondan NG, Huynh LK, Pham H, Marks M, Truong TN (2007) Macromolecules 40:4370
Sreejyothi P, Sarkar P, Dutta S, Das A, Pati SK, Mandal SK (2022) Chem Commun 58:9540
Bloom SM, Buckler SA, Lambert RF, Merry EV (1970) J Chem Soc D 870
Satpathi B, Dutta L, Ramasastry SSV (2019) Org Biomol Chem 17:1547
Zhang J, Mück-Lichtenfeld C, Studer A (2023) Nature 619:506
Thevenon A, Cyriac A, Myers D, White AJP, Durr CB, Williams CKJ (2018) J Am Chem Soc 140:6893
Blessing RH (1995) Acta Crystallogr Sect A: Found Crystallogr 51:33
Sheldrick GM (2008) Acta Crystallogr Sect A: Found Crystallogr 64:112
Momma K, Izumi F (2011) J Appl Crystallogr 44:1272
Acknowledgements
Financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development, and the Christian Doppler Research Association (Christian Doppler Laboratory for Organocatalysis in Polymerization) is gratefully acknowledged. JAH acknowledges funding by the Austrian Science Fund (FWF) via the grant https://doi.org/10.55776/P32005 and https://doi.org/10.55776/P35963. Authors thank Johanna Lang, David Edinger and Viktor Schallert for STA measurements.
Funding
Open access funding provided by Graz University of Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Steiner, M.R., Marschner, C., Baumgartner, J. et al. Synthesis, characterization and stability of phosphonium phenolate zwitterions derived from a (diphenylphosphino)phenol derivative and oxiranes. Monatsh Chem 155, 715–723 (2024). https://doi.org/10.1007/s00706-024-03216-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-024-03216-1