
Abstract
A Brønsted acid-catalyzed regioselective intramolecular hydroalkoxylation is described. This reaction proceeds via a carbocation intermediate and enables the preparation of 1,1,1’-trisubstituted tetrahydropyran derivatives under mild conditions using catalytic triflic acid (TfOH) and hexafluoroisopropanol (HFIP) as a mediator.
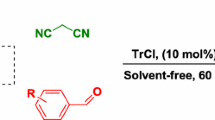
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Substituted tetrahydropyrans and tetrahydrofurans are common structural units found in a plethora of antibiotics and biologically active natural products [1], such as marine natural products [2]. Additionally, they are also present in perfumes and flavoring ingredients [3,4,5].
Given the broad applications and synthetic demand of related compounds, they have been the subject of considerable investigations in the past decades. One of the most straightforward approaches to the synthesis of cyclic ethers is the intramolecular hydroalkoxylation (or hydroetherification) using unsaturated alcohols. In this context, several methods employing a catalytic silane-iodine system [6], phenylselenium chloride [7], reductive organomercurials [8], or catalytic amounts of palladium- [9] and platinum-salts [10] have been reported.
In addition to the aforementioned transition-metal-catalyzed protocols, a method employing cationic lanthanide complexes has also been reported [11] (Scheme 1, A top). However, given the low boiling points of the products, isolation proved challenging and low isolated yields were obtained in many cases. In a similar approach, Duñach reported that Brønsted acids can also catalyze the hydroalkoxylation of various alkenes, albeit also with diminished yields due to volatility and forming mixtures of regioisomeric products [12] (Scheme 1, A bottom).

As part of our ongoing research on alkene functionalization [13] and complemented by a serendipitous discovery, we observed the hydroalkoxylation of alkenes in the presence of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP).
Whereas previously reported methods often face regioselectivity challenges, forming both 5- and 6-membered cyclic ethers as a result of the positioning of the internal double bond, in our case, the protonation of the terminal double bond allows for complete control of regioselectivity, forming exclusively the 6-membered cyclic ether products. We herein report the formation of substituted tetrahydropyrans (THPs) under Brønsted acid catalysis, mediated by HFIP (Scheme 1, B).
Results and discussion
At the outset, we observed the formation of tetrahydropyran 2a from 1a in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) in HFIP in high yield. With these initial results in hand, we investigated different solvents (Table 1, entries 1–4), and were pleased to see that reducing the amount of HFIP, using DCM as the reaction medium, allowed the obtention of 2a in comparable yield (entry 4).

Several Brønsted acids were additionally surveyed (Table 1, entries 5–7), with triflic acid providing the highest yield (91%, entry 5), while the reaction was found not to proceed in the absence of acid, even when 5.0 eq. of HFIP were used (entry 8). Varying the temperature from ambient conditions (23 °C) was determined to be detrimental to the reaction (not shown) and the amount of triflic acid (entries 9–12) was further optimized, revealing the possibility for performing this reaction as a catalytic process. Ultimately, the use of 20 mol% TfOH and 5.0 eq. of HFIP yielded the best result (entry 13, 98% NMR yield, 92% isolated).
With optimized conditions in hand, the unsaturated alcohols 1a, 1b, and 1c were subjected to the reaction protocol (Table 2). Pleasingly, the presence of side chains containing both aromatic and aliphatic substituents had no discernable influence of the efficiency of the reaction and THP derivatives 2a, 2b, and 2c were obtained in 92%, 91%, and 89% yield, respectively. Additionally, commercially available alcohol 1d was, after prolonged reaction time (14 h), fully converted and we were able to isolate the bridged bicyclic THF derivative 2d in 79% yield. In this case, we observed formation of the THF derivative instead of the corresponding THP, due to the reduced ring strain of 2d compared to its 1,4-regioisomer, the latter requiring a flip towards its less stable boat conformation (Table 2, compound 2d’). The highly volatile compound 2d has been reported to have a strong camphoraceous odour and pleasingly we were able to isolate it in 79% yield [14].
Due to their volatile nature, the isolation of low-molecular-weight ethers can be challenging [11, 12]. Therefore, we were pleased to see that, following completion of the reaction, a simple wash with 1 M sodium hydroxide and subsequent filtration through a short pad of silica gel, after careful removal of DCM under slightly reduced pressure, leads to the desired products in high yields and purity, without the need for additional purification.
We believe that the mechanism of this transformation involves the initial protonation of the double bond and formation of a tertiary carbocation (I, Scheme 2). Subsequent intramolecular attack by the hydroxyl group then leads to a protonated oxonium species (II) that, in return, gets deprotonated to form the desired cyclic ether. As shown in Table 1, the efficiency of this transformation is strongly dependent on the use of HFIP as an additive – compare entries 11 and 12. Based on previous reports suggesting that HFIP can act as a highly efficient mediator for proton-transfer processes [15,16,17,18], we believe that it assumes a similar role in the reaction reported herein, thus enabling the use of a catalytic amount of Brønsted acid.

Conclusion
We have reported a Brønsted acid-catalyzed hydroalkoxylation of unsaturated alcohols leading to highly volatile compounds under mild conditions. The facile set-up and simple isolation protocol allow the obtention of the compounds in consistently high yields and purity without the need for additional purification.
Experimental
Unless otherwise stated, all glassware was flame-dried before use and all reactions were performed under an atmosphere of argon. All solvents were distilled from appropriate drying agents prior to use or, if purchased in anhydrous form, used as received. All reagents were used as received from commercial suppliers. Reaction progress was monitored by thin layer chromatography (TLC) performed on aluminium plates coated with silica gel F254 with 0.2 mm thickness. Chromatograms were visualized by fluorescence quenching with UV light at 254 nm, or by staining using either potassium permanganate or phosphomolybdic acid. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck and co.). Neat infrared spectra were recorded using a Perkin-Elmer Spectrum 100 FT-IR spectrometer. Wavenumbers (\(\bar{v}\)) are reported in cm−1. HR–ESI–MS spectra (m/z = 50–1900) were obtained in a maXis UHR ESI-Qq-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) in the positive and/or negative ion mode by direct infusion. The sum formulas of the detected ions were determined using Bruker Compass DataAnalysis 4.1 based on the mass accuracy (Δm/z ≤ 5 ppm) and isotopic pattern matching (SmartFormula algorithm). All 1H NMR, 13C DEPTQ-135 NMR, and 13C CPD NMR spectra were recorded using a Bruker AV-400, AV-500, AV-600, or AV-700 spectrometer at 300 K. Chemical shifts are given in parts per million (ppm, δ), referenced to the solvent peak of CDCl3, defined at δ = 7.26 ppm (1H NMR) and δ = 77.16 ppm (13C NMR) [19]. Coupling constants (J) are quoted in Hz. 1H NMR splitting patterns are designated as singlet (s), doublet (d), triplet (t), or quartet (q) as they appeared in the spectrum. If the appearance of a signal differs from the expected splitting pattern, the observed pattern is designated as apparent (app). Splitting patterns that could not be interpreted or easily visualized are designated as multiplet (m) or broad (br).
General procedure for the synthesis of unsaturated alcohols
To a flame-dried Schlenk flask containing a suspension of Mg powder (30.0 mmol, 3 eq.) in dry THF (40.0 cm3) was added an iodine crystal at ambient temperature and the mixture was stirred for 10 min. Then, the corresponding bromide (20.0 mmol, 2.0 eq.) was added via syringe pump over 1 h and the reaction was kept stirring at room temperature for 12 h.
The resulting Grignard reagent was added to a suspension of the corresponding epoxide (10.0 mmol, 1.0 eq.) and CuCN (1.0 mmol, 0.1 eq.) in THF (5 cm3) at −78 °C. The reaction mixture was allowed to warm to room temperature over 12 h and a sat. aq. solution of NH4Cl (20 cm3) was slowly added. The resulting mixture was extracted with diethyl ether and the organic phase was dried over anhydrous MgSO4 and filtered. The solvent was removed under reduced pressure and the crude product was purified by flash column chromatography on silica gel.
6-Methylhept-6-en-2-ol (1a)
Following the general procedure, 1a was isolated as a colorless oil (1.10 g, 8.60 mmol, 86%) after purification by column chromatography (heptane/ethyl acetate = 5:1, Rf = 0.34). 1H NMR (400 MHz, CDCl3): δ = 4.76–4.63 (m, 2H), 3.88–3.74 (m, 1H), 2.03 (t, J = 6.4 Hz, 2H), 1.71 (s, 3H), 1.60–1.39 (m, 4H), 1.30 (d, J = 4.2 Hz, 1H), 1.20 (d, J = 6.2 Hz, 3H) ppm. All spectroscopic data was found to be in accordance with literature [20].
7-Methyl-1-phenyloct-7-en-3-ol (1b, C 15 H 22 O)
Following the general procedure, 1b was isolated as a colorless oil (304 mg, 1.39 mmol, 70%) after purification by column chromatography (heptane/ethyl acetate = 5:1, Rf = 0.29). 1H NMR (400 MHz, CDCl3): δ = 7.34–7.27 (m, 2H), 7.23–7.15 (m, 3H), 4.69 (d, J = 15.2 Hz, 2H), 3.64 (dd, J = 7.6, 4.6 Hz, 1H), 2.89–2.74 (m, 1H), 2.68 (ddd, J = 13.7, 9.5, 6.8 Hz, 1H), 2.02 (t, J = 6.3 Hz, 2H), 1.86–1.74 (m, 2H), 1.71 (s, 3H), 1.63–1.57 (m, 2H), 1.54–1.41 (m, 2H), 1.34 (t, J = 3.9 Hz, 1H) ppm; 13C NMR (101 MHz, CDCl3): δ = 145.9, 142.3, 128.6 (4C), 126.0, 110.2, 71.4, 39.3, 37.8, 37.3, 32.2, 23.7, 22.5 ppm; HRMS (ESI+): exact mass calculated for [M + Na]+ (C15H22ONa) requires m/z = 241.1563, found 241.1554; IR: \(\bar{v}\) = 3353, 3065, 3026, 2934, 2861, 1649, 1603, 1495, 1454, 1373, 1318, 1265, 1174, 1091, 1058, 1031, 969, 885, 805, 740 cm−1.
2-Methylpentadec-1-en-6-ol (1c, C 16 H 32 O)
Following the general procedure, 1c was isolated as a colorless solid (315 mg, 1.31 mmol, 66%) after purification by column chromatography (heptane/ethyl acetate = 5:1, Rf = 0.37). 1H NMR (400 MHz, CDCl3): δ = 4.69 (d, J = 11.0 Hz, 2H), 3.60 (br s, 1H), 2.03 (t, J = 7.4 Hz, 2H), 1.72 (s, 3H), 1.68–1.36 (m, 7H), 1.35–1.20 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 146.0, 110.1, 72.1, 37.9, 37.7, 37.2, 32.0, 29.9, 29.8, 29.7, 29.5, 25.8, 23.7, 22.8, 22.5, 14.3 ppm; HRMS (ESI+): exact mass calculated for [M + H]+ (C16H32OH) requires m/z = 241.2526, found 241.2517; IR: \(\bar{v}\) = 3353, 2622, 2853, 1650, 1374, 1350, 1129, 1097, 1066, 1040, 1012, 885, 721 cm−1.
General procedure for the synthesis of tetrahydropyrans (THPs) 2a-2c and tetrahydrofuran (THF) 2d
To an oven-dried vial containing 0.2 mmol of the corresponding alcohol (1 eq.), HFIP (5 eq.) and anhydrous DCM (2 cm3), TfOH (20 mol%, 40 mm3 of a 1 M solution in anhydrous DCM) was added and the reaction was stirred for 1 h at 23 °C. After consumption of the starting material as indicated by TLC analysis, a 1 M aq. solution of NaOH (0.2 cm3) was added and the resulting mixture was transferred to a separation funnel, diluted with DCM (3 cm3) and washed twice with a 1 M aq. solution of NaOH (5 cm3). The organic phase was dried over MgSO4, filtered through a short pad of silica (eluting with DCM), and carefully concentrated under reduced pressure (700 mbar, water bath temperature = 40 °C, 30 min) to give the title compound.
2,2,6-Trimethyltetrahydro-2H-pyran (2a)
Following the general procedure, 2a was isolated as a colorless oil (24 mg, 0.18 mmol, 92%). 1H NMR (400 MHz, CDCl3): δ = 3.67 (dqd, J = 12.2, 6.1, 2.2 Hz, 1H), 1.63 (ddt, J = 8.0, 7.0, 3.7 Hz, 2H), 1.54 (ddd, J = 12.8, 3.9, 2.5 Hz, 1H), 1.48–1.29 (m, 2H), 1.20 (d, J = 5.2 Hz, 6H), 1.11 (d, J = 6.1 Hz, 3H), 1.14–1.02 (m, 1H) ppm; 13C NMR (101 MHz, CDCl3): δ = 72.0, 66.6, 36.1, 33.6, 32.1, 22.8, 22.2, 20.2 ppm; HRMS (ESI+): exact mass calculated for [M + H]+ (C8H16OH) requires m/z = 129.1274, found 129.1273; IR: \(\bar{v}\) = 2972, 2930, 2866, 1459, 1445, 1379, 1364, 1347, 1339, 1281, 1236, 1214, 1191, 1154, 1138, 1089, 1061, 1005, 986, 964, 954, 842, 821 cm−1. All spectroscopic data was found to be in accordance with the literature [21].
2,2-Dimethyl-6-phenethyltetrahydro-2H-pyran (2b)
Following the general procedure, 2b was isolated as a colorless oil (40 mg, 0.18 mmol, 91%). 1H NMR (400 MHz, CDCl3): δ = 7.32–7.26 (m, 2H), 7.24–7.14 (m, 3H), 3.58–3.41 (m, 1H), 2.76 (ddd, J = 14.5, 9.3, 5.5 Hz, 1H), 2.66 (ddd, J = 13.8, 9.0, 7.4 Hz, 1H), 1.89–1.76 (m, 1H), 1.71–1.59 (m, 3H), 1.58–1.50 (m, 1H), 1.49–1.33 (m, 2H), 1.24 (s, 3H), 1.18 (s, 3H), 1.17–1.08 (m, 1H) ppm; 13C NMR (101 MHz, CDCl3): δ = 142.8, 128.7 (2C), 128.3 (2C), 125.7, 71.7, 69.3, 38.5, 36.5, 32.1, 31.8, 31.8, 22.2, 20.2 ppm; MS (QTOF, EI+, 70 eV): exact mass calculated for [M]+ (C15H22O) requires m/z = 218.1671, found 218.1662; IR: \(\bar{v}\) = 3062, 3026, 2972, 2929, 2865, 1717, 1603, 1541, 1495, 1455, 1379, 1364, 1349, 1281, 1229, 1213, 1154, 1130, 1087, 1043, 976, 931, 919, 905, 872, 854, 840, 815, 746 cm−1. All spectroscopic data was found to be in accordance with the literature [22].
2,2-Dimethyl-6-nonyltetrahydro-2H-pyran (2c, C 16 H 32 O)
Following the general procedure, 2c was isolated as a colorless oil (43 mg, 0.18 mmol, 89%). 1H NMR (400 MHz, CDCl3): δ = 3.59–3.33 (m, 1H), 1.69–1.59 (m, 2H), 1.59–1.51 (m, 1H), 1.43 (ddd, J = 6.8, 5.9, 2.5 Hz, 2H), 1.39–1.23 (m, 16H), 1.19 (s, 3H), 1.17 (s, 3H), 1.11–0.97 (m, 1H), 0.87 (t, J = 6.8 Hz, 3H) ppm; 13C NMR (101 MHz, CDCl3): δ = 71.6, 70.5, 37.2, 36.5, 32.1, 32.0, 31.7, 30.0, 29.8, 29.7, 29.5, 25.7, 22.8, 22.2, 20.3, 14.3 ppm; HRMS (GC–TOF, EI+, 70 eV): exact mass calculated for [M-H]+ (C16H31O) requires m/z = 239.2369, found 239.2364; IR: \(\bar{v}\) = 2971, 2923, 2853, 1459, 1378, 1364, 1230, 1214, 1091, 1059, 1045, 977 cm−1.
6-Oxabicyclo[3.2.1]octane (2d)
Following the general procedure, 2d was isolated as a colorless oil (35.5 mg, 0.32 mmol, 79%). 1H NMR (400 MHz, CDCl3): δ = 4.38–4.22 (m, 1H), 3.88 (d, J = 7.7 Hz, 1H), 3.81 (dd, J = 7.7, 4.5 Hz, 1H), 2.40–2.34 (m, 1H), 1.92–1.82 (m, 1H), 1.81–1.68 (m, 2H), 1.66–1.45 (m, 4H), 1.40–1.28 (m, 1H) ppm; 13C NMR (101 MHz, CDCl3): δ = 75.7, 72.6, 38.3, 35.6, 32.5, 30.3, 18.9 ppm. All spectroscopic data was found to be in accordance with the literature [23].
Data Availability
The data that support the findings of this study are available in the supplementary material of this article.
References
Boivin TLB (1987) Tetrahedron 43:3309
Lorente A, Lamariano-Merketegi J, Albericio F, Álvarez M (2013) Chem Rev 113:4567
Narula APS (2003) Perfum Flavor 28:62
Hochstetler AR (1985) Substituted tetrahydrofurans. U.S. Patent 4549029f200, Oct 22, 1985. Chem Abstr 104:115895
Noire PD (1996) Preparation of cycloalkyltetrahydrofurans and analogs as perfume fragrances. U.S. Patent 5510326, Apr 23, 1996. Chem Abstr 125:86473
Fujita S, Abe M, Shibuya M, Yamamoto Y (2015) Org Lett 17:3822
Gruttadauria M, Aprile C, Riela S, Noto R (2001) Tetrahedron Lett 42:2213
Kang SH, Lee JH, Lee SB (1998) Tetrahedron Lett 39:59
Arai MA, Kuraishi M, Arai T, Sasai H (2001) J Am Chem Soc 123:2907
Miura K, Horiike M, Inoue G, Ichikawa J, Hosomi A (2008) Chem Lett 37:270
Zhu X, Li G, Xu F, Zhang Y, Xue M, Shen Q (2017) Tetrahedron 73:1451
Coulombel L, Duñach E (2004) Green Chem 6:499
Brutiu BR, Iannelli G, Riomet M, Kaiser D, Maulide N (2024) Nature 626:92
Clarke MF, Owen LN (1950). J Chem Soc. https://doi.org/10.1039/jr9500002108
Lemmerer M, Riomet M, Meyrelles R, Maryasin B, González L, Maulide N (2022) Angew Chem Int Ed 61:e202109933
Colomer I, Chamberlain AER, Haughey MB, Donohoe TJ (2017) Nat Chem Rev 1:0088
Ratnikov MO, Tumanov VV, Smit WA (2010) Tetrahedron 66:1832
Llopis N, Baeza A (2020) Molecules 25:3464
Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI (2010) Organometallics 29:2176
Li J, Preinfalk A, Maulide N (2019) J Am Chem Soc 141:143
Dzudza A, Marks TJ (2009) Org Lett 11:1523
Francesco IN, Cacciuttolo B, Pucheault M, Antoniotti S (2014) Green Chem 17:837
Baldwin SW, Doll RJ, Haut SA (1974) J Org Chem 39:2470
Acknowledgements
This work has been supported by the Austrian Academy of Sciences (DOC Fellowship to B.R.B.) and the European Research Council (CoG VINCAT to N.M.). Generous support by the University of Vienna and by the Vienna Doctoral School in Chemistry (DoSChem) is acknowledged.
Funding
Open access funding provided by University of Vienna. HORIZON EUROPE European Research Council,CoG 682002,Nuno Maulide,Österreichischen Akademie der Wissenschaften,DOC,Bogdan Brutiu.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brutiu, B.R., Tang, L., Kaiser, D. et al. Tetrahydropyran synthesis mediated by catalytic triflic acid and hexafluoroisopropanol. Monatsh Chem 155, 709–713 (2024). https://doi.org/10.1007/s00706-024-03214-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-024-03214-3