
Abstract
Herein, we present an alternative and elegant synthetic approach toward powerful β-glucosidase inhibitor isofagomine. Derivatizations of the ring nitrogen provided a selected set of N-modified isofagomine analogues. Biological evaluation of these compounds showed a remarkable change in potency as well as α/β-preference for various glycosidases from different sources when compared to the parent compound isofagomine. Overall, the conducted N-modification improved the potency against α-glucosidase from Saccharomyces cerevisiae (GH13). Coming along, significant diminished activities toward GH1 family β-glucosidases from three different sources have been observed for all tested derivatives. Moreover, and contrary to isofagomine, deactivations of β-galactosidase from Escherichia coli (GH2) as well as α-mannosidase from Canavalia ensiformis (GH38) have not been verified for this series of compounds.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Isofagomine (1; IFG, 1,2,5-trideoxy-2-C-hydroxymethyl-1,5-imino-d-xylitol, or (3R,4R,5R)-5-(hydroxymethyl)piperidine-3,4-diol) [1], a nitrogen containing chimeric structure to d-glucose (2), has been proven as highly potent inhibitor for respective β-glucosidases (glycoside hydrolase family GH1 and GH30) [2, 3]. Therefore, this prominent parent compound of isoiminosugars (A, Fig. 1) gained profound interest in the broad field of glycosciences. Hence, developing synthetic approaches for access to suitable bench-amounts and structural modifications for tailored biological elaborations is aimed for. Accordingly, dedicated derivatizations (Fig. 1) of either the ring nitrogen (i.e., compound 4–9) [4,5,6,7,8,9,10,11,12,13,14], selected alcohols (i.e., compound 10 and 11) [15,16,17,18], or the carbon backbone (i.e., compound 12 and 13) [14, 19], as well as advanced combinations thereof (i.e., 14) [20], provided a broad set of innovative inhibitors of respective glycosidases. In consequence, appropriate structures of this compound class have been elaborated upon their biological activity against selected human glycosidases related to lysosomal storage diseases [3, 21]. In particular, Morbus Gaucher (GD, OMIM #230,800, ORPHA35) [22], the disorder caused by impaired human β-glucosidase (glucocerebrosidase, GBA, EC. 3.2.1.45, GH30) is currently the focus of various dedicated investigations [5, 57].

Structures of isofagomine (1) and selected modified derivatives (3–14; dashed box) as representatives of isoiminosugars (A) and iminosugar analogue (B) 1-deoxynojirimycin (3) and d-glucose (2)
Connected to this, guiding leaders in this field emphasized isofagomine (1) and analogues for their frequently strong complexation by general acid/base and catalytic nucleophilic amino acid residues (aspartic and/or glutamic acids) found in the active site of certain glycosidases [15, 16, 23,24,25]. Thereby, and contrasting the general behavior of related iminosugars (B), such isoiminosugars (A) interact with these two acid residues working as a “clamp” or “pincer” complexing the ring nitrogen and consequently exerting very powerful coordination (see Fig. 2).

Coordination studies based on co-crystals of a inhibitor 3 and b IFG (1), respectively, with β-glucosidase from Thermotoga maritima (T. mar., GH1). Taken and rearranged from [25]
This interesting feature of double coordination of the protonated inhibitor species with both active site residues can be seen as consequence of the inhibitors’ structural characteristics. Accordingly, isofagomine (1) is not only unique in terms of active site binding but also poses synthetic challenges when compared to related iminosugars such as 1-deoxynojirimycin (3; 1,5-dideoxy-1,5-imino-d-glucitol, or (2R,3R,4R,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol) [26]. Thus, respective C–C bond formation combined with the replacement of the anomeric carbon by a nitrogen atom is required en route toward this carbon chain branched 2,5-dideoxygenated derivative of d-glucose (2).
The first synthesis of parent compound isofagomine (1) was reported by Lundt, Bols [1] and their co-workers taking advantage of the “Černý epoxide” [27] as their crucial intermediate. Valuable alternatives based on monosaccharidic starting materials were added to the repertoire by the groups of Bols [28], as well as by Stick [4, 29, 30]. Another sugar-based approach was published by Chmielewski who provided the (-) enantiomer [31]. In addition, a range of de novo syntheses were published starting, for example, from dihydrouracil [32], from butadiene monoxide [33,34,35], from enantiomerically enriched tris(hydroxymethyl)methane (2-hydroxymethyl-1,3-dihydroxypropane) [36, 37], and from chiral (lipase-resolved) N-BOC-5-hydroxy-3-piperidene [38]. Parent compound 1 was also accessed by intramolecular delivery of a d-tartaric acid-derived α-trimethylsilylmethylamine radical cation to a tethered acetylene moiety [39, 40], or starting from arecoline [41] and related intermediates [42, 43], as well as from methyl nicotinate [44].
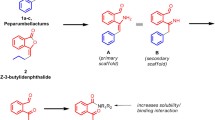
Based on the indicated importance and exceptional biological properties of isofagomine (1) and related derivatives, complementing investigations connected to respective inhibition properties toward additional glycoside hydrolases, and in consequence a convenient access to suitable bench-amounts of the parent compound, can be deemed crucial. In this context, we herein present an adapted synthetic pathway starting from d-arabinose (15; see Scheme 1) inspired by “An Efficient” [28] as well as “Expeditious” [30] synthesis of isofagomine (1) provided by the teams of Bols and Stick, respectively. Starting from cheap and commercially available carbohydrate 15, compound 18 was easily available by similar [30] and concise protecting group manipulations. Thus, initially selective access to position C-4 was gained (Scheme 1). In contrast to the following nucleophilic addition of a cyanide ion, we herein took advantage of a well-established [45,46,47] Henry reaction-based approach to simultaneously introduce a nitrogen atom combined with structurally required carbon chain branching at this position [28]. Accordingly, convenient Swern oxidation of alcohol 18 provided the respective ulose 19, as suitable and analogues intermediate for the subsequent addition of nitromethane. As consequence of the sterically more demanding character of the herein globally O-protected compound 19, this reaction turned out as highly stereoselective. Subsequently, similar and much to our delight also stereo controlled conversions of the resulted crystalline Henry-product 20 have been tested for an optimized access toward IFG (1). Thus, simple and straight forward, in this approach, virtually stereo selective as well as high yielding reactions enabled a convenient access to IFG (1) in nine steps and 35% overall yield. In the following, we prepared a selected set of differently N-modified isofagomine derivatives (27–35) for in-depth biological evaluations with a series of different glycoside hydrolases. Two types of spacer arms providing a terminal azide functionality were introduced by simple N-alkylation reactions. To increase the water solubility of the final compounds, the first series of inhibitors provides an “ethylene glycol” based spacer arm (Scheme 2, series A). Their corresponding counterparts exhibit a simple alkyl chain (Scheme 2, series B), thereby targeting lipophilic interactions with respective areas adjacent to the active site of enzymes investigated herein. Subsequent reductive liberation of the primary amines 29 and 30 enabled concise conversions to selected sulfonamides, inhibitors 31–35 (see Scheme 3).



Results and discussion
In detail, starting from crystalline (see Fig. 3) benzyl 4-O-acetyl-β-d-arabinopyranoside (16) [30], treatment with dimethoxymethane, acetyl chloride (AcCl), and catalytic amounts of zinc chloride (ZnCl2) in dichloromethane (CH2Cl2) [48] provided global protected compound 17 in 88% (see Scheme 1). Subsequent liberation of OH-4 under Zemplen’s conditions yielded alcohol 18, which in turn was oxidized to ulose 19 employing Swern’s methodology (93%, over 2 steps).

Crystal structures of: A compound 16 (CCDC 2330318) and B compound 20 (CCDC 2330319), respectively
In a well-established synthetic procedure [45,46,47], including a common Henry reaction, followed by sequential activation and elimination sequence, the corresponding condensation product 22 was formed. In detail, compound 19 was treated with nitromethane (MeNO2) and potassium tert-butoxide (t-BuOK) in tetrahydrofuran (THF) yielding the Henry product 20 (81% starting from 18). It is noteworthy to mention, that completed conversion of the starting material was already detected after stirring for 5 min at ambient temperature. To our delight, compound 20 was formed as virtually single and crystalline diastereomer confirmed by NMR analysis (4S/4R approx. 6/1 determined by NMR). Thereby, the nucleophilic attack predominantly (compare reiterated NMR-studies of corresponding crude reaction mixtures; Fig. SI-16a) occurred from Ré-face, which was structurally unambiguously confirmed by XRD-studies (see Fig. 3).
Subsequently, acid-catalyzed esterification of alcohol 20 in acetic anhydride (Ac2O) provided compound 21 in 86% yield. Concomitantly, the methoxymethyl (MOM) protecting groups at positions O-2 and O-3 were converted into acetyloxymethylene groups. Assumed issues regarding chemical stability of these adapted protecting groups (coincident lability against acids as well as bases) could not have been verified in our hands, compound 21 turned out as highly bench stable. Moreover, stirring at T = 80 °C in toluene for 3 days only provided insignificant amounts of nitroalkene 22 and did not lead to any other decomposition of the starting material 21. Even treatment with excess amounts of sodium borohydride (NaBH4) in MeOH showed no influence on the protecting groups. Accordingly, the formal elimination of AcOH was induced by treatment of intermediate 21 with NaHCO3 in THF at T = 40 °C (see Scheme 1). Following this, complete conversion of the starting material was already obtained after 3 h in 83%. Thereby, predominantly the (E) configurated stereoisomer of product 22 (E/Z approx. 8/1, in extended NMR-experiments (APT, NOE); see Fig. SI-16b and SI-16c) was formed. To optimize the final cyclization-deprotection sequence leading to IFG (1), different synthetic strategies (see Scheme 1) starting from nitroalkene 22 were tested. Initial cleavage of the protecting groups under strong acidic conditions, followed by catalytic hydrogenolysis (entry A) was not successful in our hands. Next, treatment with hydrogen in methanolic HCl over Pd/C (entry B) provided a mixture of isofagomine (1) and its C-5 epimer 24 in a ratio about 2:1, as well as several unidentified side products. In consequence, unsatisfying isolated yields (max. 20% of 1) verified this entry B as not convenient for us. In contrast, initial stirring of compound 22 under an atmosphere of hydrogen over Pd(OH)2/C in MeOH until complete reductive cyclization, followed by subsequent treatment with methanolic HCl (3 N), afforded free inhibitors 1 (71%) and 24 in a ratio of approximately 3:1 (entry C). Following entry D, this desired steric outcome was significantly increased by precedent reduction of the double bond with sodium borohydride (NaBH4) in MeOH, thereby obtaining the corresponding intermediate 23. Triggered by lowered reaction temperatures, this stereoselective 1,4-addition of the hydride ion, predominantly formed the (4R) configurated diastereomer (compare summarized NMRs of the respective crude reaction mixtures in Fig. SI-16d). Subsequent catalytic hydrogenolysis of intermediate 23 under strong acidic conditions in MeOH resulted in global O-deprotection and reduction of the nitro moiety. Concomitant cyclization in an intramolecular reductive amination reaction yielded the desired free IFG (1) in 88% starting from 22 (see Scheme 1).
For N-modification (Scheme 2), IFG (1) was treated with 1-azido-2-[2-(2-bromoethoxy)ethoxy]ethane (25) [49] in the presence of trimethylamine (Et3N) in dimethylformamide (DMF) at T = 70 °C to provide compound 27 in 60% yield. Accordingly, N-alkylated analogue 28 (66%) was obtained by conversion with 1-azido-6-bromohexane (26) [50]. Catalytic reduction of the respective azides in MeOH provided the corresponding amines 29 (91% yield from 27) and 30 (85% yield from 28), respectively.
In the following, common conversions of amines 29 and 30 with different sulfonyl chlorides and Et3N in MeOH provided the corresponding sulfonamides 31–35 (see Scheme 3). In detail, conversions with 4-methylbenzene-1-sulfonyl chloride (“tosyl” chloride; TsCl) yielded the electron-rich aromatic sulfonamides 31 and 32 in 75% and 71%, respectively. The electron-poor aromatic system was provided by the formal exchange of the involved methyl group (+ I effect) by a bromo substituent (-I effect). Thus, amine 30 was treated with 4-bromobenzene-1-sulfonyl chloride (“brosyl” chloride; BsCl) to give the corresponding NHbrosyl derivative 34 in 72%. As consequence of longstanding experiences with inhibitors providing 5-(dimethylamino)naphthalene-1-sulfonamides (“dansyl”), known to significantly increase biological activities with respective enzymes [51,52,53,54,55], we herein wanted to take advantage of equal functional modification. Accordingly, fluorescently tagged compounds 33 and 35 were synthesized via conversions of amines 29 and 30 with 5-(dimethylamino)naphthalene-1-sulfonyl chloride (“dansyl” chloride; dansylCl) in 65% and 58% yield, respectively.
Biological evaluation
All title compounds 27–35 were evaluated concerning their inhibitory activities against several glycoside hydrolases from various sources. In Table 1, the respective inhibition constants are collected and compared with parent compound isofagomine (1).
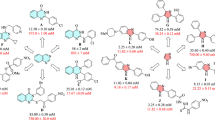
Series A inhibitors 27, 31, and 33, N-derivatives with ethylene glycol spacer arms, showed a significant loss (up to 5700-fold) of activity toward tested GH1 family β-glucosidases when compared to parent compound IFG (1). In relation to this, according deactivation potencies toward α-glucosidase from Saccharomyces cerevisiae (S. cer., GH13) were only slightly reduced (by factors of 2–6). However, amino derivative 29 provided a relatively stronger affinity toward tested β-glucosidases when compared to its corresponding “terminal nonpolar” analogues 27, 31, and 33. Compared to all inhibitors presented herein, a considerably decreased inhibition of α-glucosidase from S. cer. (GH13) was observed for 29 (Ki = 1154 µM vs. corresponding Ki-values ranging from 1.8 to 221 µM). Except relatively high potencies toward GH1 family β-glucosidases, related influences on the respective interactions with the remaining probed enzymes were not observed for corresponding amino derivative 30 from Series B. In contrast, investigations with the more lipophilic alkyl chain-based derivatives 28, 32, 34, and 35 from Series B showed a remarkable change in the potency and α/β-preference. These compounds showed significantly improved (by factors of 4–20) activities against α-glucosidase from S. cer. (GH13). In this context, our best candidate (NHbrosyl derivative 34, Ki = 1.8 µM) effectively provided a 20-fold increased potency compared to IFG (1, Ki = 35.4 µM). Simultaneously, an impressively diminished inhibition of β-glucosidases from Thermotoga maritima (T. mar.), Prunus dulcis (P. dul.) and β-glucosidase/β-galactosidase from Agrobacterium sp. (Abg), members of the GH1 family, was observed (up to 4200-fold decreased activity) with the mentioned inhibitors 28, 32, 34, and 35. In this context, supporting depictions of the presented Ki-values are provided in Fig. SI-17a and Fig. SI-17b This observed trend regarding inhibition potency of the presented compounds is additionally indicated in Fig. 4, in which respective activities of each compound with probed enzymes are normalized and depicted as relatives (f = Ki isofagomine/Ki compound) to their parent compound 1.

Relative inhibition activities of inhibitors 27, 28, and 31–35 compared to parent compound IFG (1) (f = Ki IFG/Ki compound)
This trend in α/β-preference is additionally indicated in Fig. SI-17c with Fsel = Ki β-glucosidases/Ki α-glucosidase for respective inhibitors. In addition, and contrary to isofagomine (1), all presented inhibitors 27–35 showed no mentionable deactivation of β-galactosidase from Escherichia coli (E. coli, GH2) as well as α-mannosidase from Canavalia ensiformis (C. ens., GH38).
Conclusion
We herein present an adapted, alternative synthesis of isofagomine (1) starting from cheap d-arabinose (15). Following a versatile protecting group strategy, selective access toward position C-4 was gained. Subsequent oxidation of the accessible alcohol, followed by consecutive treatment with nitromethane, provided predominantly one crystalline diastereomer of the corresponding “Henry-products”, compound 20. Conversion to nitroalkene 22, followed by distinct reduction and cyclization methods, provided isofagomine 1 in nine steps and 35% overall yield. Conventional N-alkylation of inhibitor 1, followed by derivatizations of the introduced spacer arms, yielded a set of N-modified isofagomine derivatives 27–35.
Compared to parent compound 1, all tested compounds provided significantly reduced inhibition activities of GH1 family β-glucosidases from Thermotoga maritima (T. mar.), Prunus dulcis (P. dul.) and β-glucosidase/β-galactosidase from Agrobacterium sp. (Abg). Simultaneously, an improved inhibitory potency against α-glucosidase from Saccharomyces cerevisiae (S. cer., GH 13) was observed for N-alkyl modified inhibitors 28, 32, 34, and 35 when compared to IFG (1). In addition, and contrary to IFG (1), all presented N-derivatives provided herein showed no mentionable deactivation of β-galactosidase from E. coli (GH2) as well as α-mannosidase from Canavalia ensiformis (C. ens. (GH38). Possibly, these findings help in the development of novel compounds selectively interacting with (impaired) α-glucosidases, clinically relevant for a potential treatment of Pompe disease.
Experimental
Optical rotations were measured at 20 °C using a Perkin Elmer 341, or Schmidt&Haensch VariPol C101R-NMPVS polarimeter at 589 nm with a path length of 10 cm. \({\left[\alpha \right]}_{{\text{D}}}^{20}\) values are given in 10−1 deg cm2 g−1. NMR spectra were recorded on a Bruker Ultrashield spectrometer at 75.53 MHz (13C) and 300.36 MHz (1H), respectively. CDCl3 was employed for protected compounds and CD3OD as well as D2O for unprotected inhibitors. Chemical shifts are listed in δ employing residual, non-deuterated solvent or residual H2O (CD3OD) as the internal standard. CDCl3: 7.26 ppm (1H), 77.16 ppm (13C); CD3OD: 4.87 ppm (1H), 49.0 ppm (13C); D2O: 4.79 ppm (1H). The signals were unambiguously assigned by COSY (correlation spectroscopy) and HSQC (heteronuclear single-quantum correlation spectroscopy) analysis. For all structures, the respective numbering of each atom is indicated in the supporting information. MALDI-TOF was performed on a Micromass TofSpec 2E Time-of-Flight mass spectrometer. High-resolution MS data were recorded using LC-TOF instrument (Agilent Technologies 6230 TOF LC/MS) equipped with an electrospray ionization source (ESI). Accurate mass determination was corrected by calibration using ESI-L Low Concentration Tuning Mix by Agilent Technologies. All reactions were monitored by thin-layer chromatography (TLC) performed on pre-coated aluminum plates with silica gel 60 F254 and detected with UV light (254 nm). For staining, a solution of vanillin (9 g) in a mixture of H2O/EtOH/H2SO4 (950 cm3–750 cm3–120 cm3) or ceric ammonium molybdate ((NH4)6Mo7O24·4H2O (100 g)/Ce(SO4)2·4H2O (8 g) in 10% H2SO4 (1000 cm3)) were employed, followed by heating on a hotplate.
General procedure A (hydrogenolysis over Pearlman’s catalyst)
A 10% solution of the respective starting material in MeOH was stirred with Pd(OH)2/C under an atmosphere of H2 at ambient pressure. After completed conversion of the starting material, the catalyst was filtered off and the solvent was removed under reduced pressure.
General procedure B (N-alkylation)
A 5% solution of the respective amine in dry DMF was stirred with Et3N (2.5–3.0 equiv.) and the selected halocarbon (1.1 equiv.) at T = 70 °C. After completed conversion of the starting material, the solvent was removed under reduced pressure to give the crude product.
General procedure C (sulfonamide formation)
To a 10% solution of the respective amine and Et3N (2.5–3.0 equiv.) in MeOH, the corresponding sulfonyl chloride (1.2 equiv.) was added. After completed conversion of the starting material, the reaction mixture was evaporated to dryness.
Benzyl 4-O-acetyl-2,3-di-O-methoxymethylene-β-d-arabino-pyranoside (17, C18H26O8)
To a stirred dispersion of dimethoxymethane (15.7 cm3, 214.6 mmol) and catalytic amounts of zinc chloride (48 mg, 0.35 mmol) in CH2Cl2 (250 cm3), acetyl chloride (15.3 cm3, 214.6 mmol) was added dropwise. After stirring for 4 h, a solution of compound 16 (10.1 g, 35.7 mmol) and i-Pr2EtN (74.9 cm3, 129.3 mmol) in CH2Cl2 (10 cm3) was added dropwise. After stirring for additional 16 h, the reaction mixture was consecutively washed with HCl (2 N) and saturated aqueous NaHCO3. Removal of the dried (Na2SO4) solvent under reduced pressure followed by silica gel chromatography (cyclohexane/EtOAc, 8:1 v/v) provided compound 17 (11.7 g, 31.6 mmol, 88.3%) as a pale yellow oil. Rf = 0.4 (CH/EA, 2:1 v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= − 123.6 (MeOH, c = 1.10); 1H NMR (300 MHz, CDCl3): δ = 7.42–7.22 (m, 5H, aromatic), 5.26 (m, 1H, H-4), 5.02 (d, 1H, J1-2 = 3.6 Hz, H-1), 4.79–4.51 (m, 6H, 4x –OCH2OCH3, 2x –OCH2Ph), 4.10 (dd, 1H, J2,3 = 10.2 Hz, J3,4 = 3.6 Hz, H-3), 3.92 (dd, 1H, H-2), 3.88–3.80 (m, 1H, H-5a), 3.62 (dd, 1H, J5a,5b = 12.8 Hz, J4,5b = 2.0 Hz, H-5b), 3.33 (s, 3H, –OCH2OCH3), 3.24 (s, 3H, –OCH2OCH3), 2.10 (s, 3H, CH3CO–) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 169.7 (carbonyl), 136.5 (ipso aromatic), 127.7, 127.6, 127.2 (aromatic), 96.8 (C-1), 96.7, 95.5 (2x –OCH2OCH3), 73.9 (C-2), 71.3(C-3), 69.7(C-4), 68.7 (–OCH2Ph), 60.0 (C-5), 54.8, 54.6 (2x –OCH2OCH3), 20.4 (CH3CO–) ppm; MS (MALDI-TOF): m/z calculated for [C18H26O8Na] ([M + Na]+) 393.1525, found 393.1524.
Benzyl 2,3-di-O-methoxymethylene-β-d-arabinopyranoside (18, C16H24O7)
To a 10% solution of compound 17 (6.2 g, 16.7 mmol) in MeOH, catalytic amounts of NaOMe were added to adjust pH 12. After completed conversion of the starting material, the reaction mixture was neutralized by addition of strongly acidic ion-exchange resin (Amberlite IR-120H+). After stirring for 5 min, filtration and evaporation of the solvent, the crude product was purified by silica gel chromatography (cyclohexane/EtOAc, 10:1 v/v) to give compound 18 (5.2 g, 15.8 mmol, 94.6%) as a pale yellow syrup. Rf = 0.3 (CH/EA, 1:1 v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)=− 110.0 (MeOH, c = 1.30); 1H NMR (300 MHz, CDCl3): δ = 7.44–7.23 (m, 5H, aromatic), 5.01 (d, 1H, J1,2 = 3.2 Hz, H-1), 4.85–4.49 (m, 6H, 4x –OCH2OCH3, 2x –OCH2Ph), 4.05–3.90 (m, 3H, H-2, H-3, H-4), 3.82 (d, 1H, J5a,5b = 12.5 Hz, J4,5a < 1 Hz, H-5a), 3.69 (d, 1H, H-5b), 3.38 (s, 3H, –OCH2OCH3), 3.24 (s, 3H, –OCH2OCH3), 2.95 (bs, 1H, –OH) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 137.1 (ipso aromatic), 128.2, 128.0, 127.8 (aromatic), 97.3 (C-1), 97.1, 96.6 (2x –OCH2OCH3), 74.9(C-4), 74.2 (C-2), 69.1 (–OCH2Ph), 68.5 (C-3), 61.9 (C-5), 55.5, 55.1 (2x –OCH2OCH3) ppm; MS (MALDI-TOF): m/z calculated for [C16H24O7Na] ([M + Na]+) 351.1420, found 351.1833.
Benzyl 4-C-nitromethyl-2,3-di-O-methoxymethylene-α-l-xylopyranoside (20, C17H25NO9)
To a solution of DMSO (3.0 cm3, 42.8 mmol) in 25 cm3 CH2Cl2, oxalyl chloride (2.7 cm3, 32.1 mmol) was added dropwise at T = − 78 °C. After stirring for 30 min, alcohol 18 (5.0 g, 15.3 mmol, dissolved in 25 cm3 CH2Cl2) was added dropwise and the reaction mixture was stirred for 20 min. Addition of Et3N (11.6 cm3, 84.0 mmol) and stirring for additionally 20 min completed the conversion of the reactants. The reaction mixture was consecutively washed with HCl (2 N) and saturated aqueous NaHCO3. Removal of the dried (Na2SO4) solvent under reduced pressure gave the crude ulose 19 (4.9 g) which was immediately employed in the next step without further purification. [Crude compound 19 is practically stable at T = − 18 °C for ca. 1 week. However, purification on silica gel induces undesired enolization/epimerization reactions.] Rf = 0.4 (CH/EA, 1:1 v/v).
To a freshly prepared suspension of MeNO2 (11.5 cm3, 214.6 mmol) and t-BuOK (1.7 g, 15.3 mmol) in THF (30 cm3), crude compound 19 (calculated for 15.3 mmol of alcohol 18, dissolved in 20 cm3 THF) was dropwise added. After TLC indicated completed conversion of the starting material (ca. 15 min), the reaction mixture was diluted with CH2Cl2 and consecutively washed with HCl (2 N) and aqueous saturated NaHCO3. [Longer reaction times induced the formation of additional products; most likely resulted from Retro-Henry and/or connected isomerization reactions.] Subsequently, the combined organic layers were dried over Na2SO4, filtered off and evaporated to give crude compound 20 (please compare Supporting Information Fig. SI-16a) as a white solid. Purification on silica gel (cyclohexane/EtOAc, 5:1 v/v) provided product 20 (4.7 g, 12.13 mmol, 79.4%) as a colorless crystals. Re-crystallization from cyclohexane/EtOAc (approx. 7:1 v/v) enabled XRD studies for the unambiguous structural assignment of compound 20. Rf = 0.5 (CH/EA, 1:1 v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 27.3 (EA, c = 1.28); 1H NMR (300 MHz, CDCl3): δ = 7.35–7.21 (m, 5H, aromatic), 4.89 (d, 1H, J1,2 = 3.8 Hz, H-1), 4.76–4.46 (m, 6H, 4x –OCH2OCH3, 2x –OCH2Ph), 4.64 (s, 2H, H-4′), 3.87 (d, 1H, J5a,5b = 11.9 Hz, H-5a), 3.81 (d, 1H, J2,3 = 9.9 Hz, H-3), 3.66 (d, 1H, H-5b), 3.46 (s, 3H, –OCH2OCH3), 3.33 (dd, 1H, H-2), 3.17 (s, 3H, –OCH2OCH3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 136.6 (ipso aromatic), 128.6, 128.6, 128.3 (aromatic), 99.2, 97.3 (2x –OCH2OCH3), 96.2 (C-1), 84.7 (C-3), 76.6 (C-4′), 74.6(C-2), 71.7 (C-4), 69.5 (–OCH2Ph), 61.3 (C-5), 56.7, 55.6 (2x –OCH2OCH3) ppm; MS (ESI-TOF): m/z calculated for [C17H25NO9Na] ([M + Na]+) 410.1427, found 410.1411; m.p.: 114–116 °C (recrystallized from ethyl acetate and cyclohexane).
Benzyl 4-O-acetyl-4-C-nitromethyl-2,3-di-O-acetoxymeth-ylene-alpha-l-xylopyranoside (21, C21H27NO12)
Compound 20 (3.50 g, 9.03 mmol) was dissolved in 35 cm3 Ac2O. After addition of catalytic amounts of para-toluenesulfonic acid (pTSA), the reaction mixture was stirred for 18 h. After consecutive addition of crushed ice cubes and (ice-cooled) water (ca. 100 cm3) under vigorous stirring (for 30 min), the reaction mixture was diluted with CH2Cl2. Two additional washing steps of the aqueous phase with CH2Cl2 and subsequent neutralization of the organic layer via extraction with saturated aqueous NaHCO3 provided crude product 21. Purification on silica gel (cyclohexane/EtOAc, 10:1 v/v) gave product 21 (3.8 g, 7.83 mmol, 86.6%) as a colorless syrup. Rf = 0.8 (CH/EA, 1:1 v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= − 83.1 (EA, c = 0.92); 1H NMR (300 MHz, CDCl3): δ = 7.35–7.21 (m, 5H, aromatic), 5.26 (m, 2H, –OCH2O(CO)CH3), 5.16 (m, 1H, –OCH2O(CO)CH3), 5.05 (m, 1H, –OCH2O(CO)CH3), 5.03 (d, 1H, J4′a,4′b = 12.1 Hz, H-4′a), 4.84 (d, 1H, H-4′b), 4.82 (d, 1H, H-1), 4.73 (d, 1H, J = 12.4 Hz, –OCH2Ph), 4.53 (m, 2H, H-3, –OCH2Ph), 4.12 (d, 1H, J5a,5b = 12.0 Hz, H-5a), 4.05 (d, 1H, H-5b), 3.66 (dd, 1H, J1,2 = 3.6, J2,3 = 9.4 Hz, H-2), 2.06 (s, 3H, –OCH2O(CO)CH3), 2.04 (s, 3H, –OCH2O(CO)CH3), 1.89 (s, 3H, –O(CO)CH3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 170.4, 170.2, 170.1 (3x –O(CO)CH3), 136.6 (ipso aromatic), 128.7, 128.2, 128.0 (aromatic), 95.9 (C-1), 89.0 (2x –CH2O(CO)CH3), 80.1 (C-4), 78.2 (C-3), 77.5 (C-2), 73.3 (C-4′), 69.7 (–OCH2Ph), 60.2 (C-5), 21.9, 21.1, 21.0 (3x –O(CO)CH3) ppm; MS (ESI-TOF): m/z calculated for [C21H27NO12NH4] ([M + NH4]+) 503.1912, found 503.1865.
(E)-Benzyl 4-deoxy-4-C-nitromethylene-2,3-di-O-ethanoyl-methylene-β-d-arabinopyranoside (22, C19H23NO10)
Compound 21 (3.7 g, 7.62 mmol) was dissolved in 40 cm3 THF and stirred for 10 h at T = 45 °C after addition of catalytic amounts of NaHCO3. Subsequently, the solvents were removed under reduced pressure to provide crude compound 22 (please compare Supporting Information Fig. SI-16b). Purification on silica gel (cyclohexane/EtOAc, 10:1 v/v) gave product 22 (2.70 g, 6.35 mmol, 83.3%) as a pale yellow syrup. Rf = 0.7 (CH/EA, 3:2 v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= − 251.6 (EA, c = 1.13); 1H NMR (300 MHz, CDCl3): δ = 7.35–7.26 (m, 5H, aromatic), 7.11 (s, 1H, H-4′), 5.39–5.22 (m, 3H, –OCH2O(CO)CH3), 5.14 (d, 1H, H-5a), 5.09 (d, 1H, –OCH2O(CO)CH3), 4.95 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.75 (d, 1H, J = 12.3 Hz, –OCH2Ph), 4.66 (d, 1H, H-3), 4.56 (d, 1H, –OCH2Ph), 4.25 (d, 1H, J5a,5b = 14.8 Hz, H-5b), 3.70 (dd, 1H, J2,3 = 9.8 Hz, H-2), 2.03 (s, 3H, –OCH2O(CO)CH3), 1.90 (s, 3H, –OCH2O(CO)CH3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 170.5, 170.2 (2x –O(CO)CH3), 145.3 (C-4), 136.6 (ipso aromatic), 134.9 (C-4′), 128.7, 128.3, 128.1 (aromatic), 96.2 (C-1), 89.2, 88.7 (2x –OCH2O(CO)CH3), 81.2 (C-2), 76.0 (C-3), 69.8 (–OCH2Ph), 56.7 (C-5), 21.0 (2x –OCH2O(CO)CH3) ppm; MS (ESI-TOF): m/z calculated for [C19H23NO10NH4] ([M + NH4]+) 443.1701, found 443.1646.
4-Deoxy-4-C-nitromethyl-2,3-di-O-ethanoylmethylene-β-d-arabinopyranoside (23, C19H25NO10)
To a solution of compound 22 (2.19 g, 5.15 mmol) in MeOH (30 cm3), NaBH4 (1.29 g, 33.97 mmol) was carefully added at T = − 10 °C. After TLC indicated completed conversion of the starting material (ca. 5–10 min), the reaction mixture was neutralized by addition of acidic ion-exchange resin (amberlite IR-120H+). Subsequently, filtration and evaporation of the solvent followed by purification on silica gel (cyclohexane/EtOAc, 15:1 v/v) provided a mixture of compound 23 (2.05 g, 4.80 mmol, 93.2%) and traces of an inseparable unidentified species (most likely an isomeric analogue of 23) as a white wax. Rf = 0.6 (CH/EA, 3:2 v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= − 222.0 (CHCl3, c = 1.56); 1H NMR (300 MHz, CDCl3): δ = 7.35–7.18 (m, 5H, aromatic), 5.39 (d, 1H, J = 6.5 Hz, –OCH2O(CO)CH3), 5.30 (d, 1H, J = 6.5 Hz, –OCH2O(CO)CH3), 5.09 (m, 2H, 2x –OCH2O(CO)CH3), 4.82 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.70 (d, 1H, J = 12.3 Hz, –OCH2Ph), 4.59 (dd, 1H, J4,4′a = 3.7, J4′a,4′b = 13.1 Hz, H-4′a), 4.48 (m, 2H, 1x–OCH2Ph, H-4′b), 4.12 (dd, 1H, J2,3 = 9.4, J3,4 = 5.5 Hz, H-3), 3.87 (dd, 1H, J4,5a = 2.8, J5a,5b = 12.3 Hz, H-5a), 3.50 (dd, 1H, H-2), 3.45 (dd, 1H, J4,5b = 9.2 Hz, H-5b), 2.92 (m, 1H, H-4), 2.03 (s, 3H, –OCH2O(CO)CH3), 1.89 (s, 3H, –OCH2O(CO)CH3) ppm; 13C NMR (75.5 MHz, APT, CDCl3): δ = 170.5, 170.5 (2x –O(CO)CH3), 136.9 (ipso aromatic), 128.6, 128.1, 128.1 (aromatic), 89.1 (C-1), 89.2, 88.7 (2x –OCH2O(CO)CH3), 76.5 (C-2), 76.3 (C-3), 58.9 (C-4′), 69.6 (–OCH2Ph), 58.9 (C-5), 39.5 (C-5), 21.0 (2x –OCH2O(CO)CH3) ppm; MS (ESI-TOF): m/z calculated for [C19H25NO10NH4] ([M + NH4]+) 445.1857, found 445.1823.
1,2,5-Tridexy-2-C-hydroxymethyl-1,5-imino-d-xylitol; or (3R,4R,5R)-5-(hydroxymethyl)piperidine-3,4-diol; or isofagomine (1)
Entry C: Compound 22 (0.81 g, 1.90 mmol) was dissolved in 15 cm3 MeOH and stirred over catalytic amounts of Pd(OH)2/C under an atmosphere of H2. After 18 h, 2 cm3 methanolic HCl (3 N) were added and the reaction mixture was stirred for additional 12 h. Subsequently, the catalyst was filtered off and the solvent was removed under reduced pressure. Purification on silica (CHCl3/MeOH/NH4OH (conc.), 8:1:1 v/v/v) provided compound 24 (49 mg, 0.34 mmol, 17.9%) and isofagomine (1, 201 mg, 1.36 mmol, 71.4%) as a white solid, respectively.
Entry D: To a solution of compound 22 (0.85 g, 1.99 mmol) in MeOH (10 cm3), NaBH4 (0.45 g, 11.98 mmol) was carefully added at T = − 10 °C. After TLC indicated completed conversion of the starting material (ca. 5–10 min), the reaction mixture was neutralized by addition of acidic ion-exchange resin (amberlite IR-120H+). Subsequently, filtration and evaporation of the solvent provided crude intermediate 23 (please compare Supporting Information Fig. SI-16d) which was directly dissolved in a mixture of 10 cm3 MeOH and 2 cm3 methanolic HCl (3 N) and stirred over catalytic amounts of Pd(OH)2/C under an atmosphere of H2 for 16 h. Subsequently, the catalyst was filtered off and the solvent was removed under reduced pressure. Purification on silica provided title compound isofagomine (1, 260 mg, 1.77 mmol, 88.4%) as a white solid, respectively. NMR data fits to the literature [1].
(3R,4R,5R)-1-[2-[2-(2-Azidoethoxy)ethoxy]-ethyl]-5-(hydroxymethyl)piperidine-3,4-diol; or N-(8-azido-3,6-dioxaoctyl)isofagomine (27, C12H24N4O5)
Following general procedure B, compound 1 (121 mg, 0.815 mmol) was treated with 1-azido-2-[2-(2-bromoethoxy)ethoxy]ethane (25, 214 mg, 0.897 mmol) [49]. Purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v) provided N-alkylated derivative 27 (160 mg, 0.532 mmol, 64.5%) as a colorless syrup. Rf = 0.5 (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 10.0 (MeOH, c = 1.10); 1H NMR (300 MHz, D2O): δ = 3.81 (dd, 1H, J5,6a = 3.4, J6a,6b = 11.4 Hz, H-6a), 3.77–3.66 (m, 8H, 2 × H-2′, 2 × H-3′, 2 × H-4′, 2 × H-5′), 3.65–3.57 (m, 2H, H-3, H-6b), 3.51 (t, 2H, J = 4.9 Hz, 2 × H-6′), 3.24 (dd, 1H, J3,4 = J4,5 = 9.8 Hz, H-4), 3.12 (dd, 1H, H-2eq), 3.06 (dd, 1H, H-5aeq), 2.70 (t, 2H, J = 5.6 Hz, 2 × H-1′), 2.08 (dd, 1H, J5aax,5aeq = J5,5aax = 11.9 Hz, H-5aax), 2.07 (dd, 1H, J2ax,2eq = J2ax,3 = 11.0 Hz, H-2ax), 1.77 (m, 1H, H-5) ppm; 13C NMR (75.5 MHz, D2O): δ = 73.7 (C-4), 71.1 (C-3), 69.6, 69.4, 69.2, 67.6 (C-2′, C-3′, C-4′, C-5′), 60.7 (C-6), 57.5 (C-2), 56.0 (C-1′), 54.5 (C-5a), 50.2 (C-6′), 42.7 (C-5) ppm; MS (ESI-TOF): m/z calculated for [C12H24N4O5H] ([M + H]+) 305.1825, found 305.1835.
(3R,4R,5R)-1-[2-[2-(2-Aminoethoxy)ethoxy]-ethyl]-5-(hydroxymethyl)piperidine-3,4-diol; or N-(8-amino-3,6-dioxaoctyl)isofagomine (29, C12H26N2O5)
According to general procedure A, compound 27 (122 mg, 0.394 mmol) was converted into its corresponding crude amine 29. Subsequent purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v) provided compound 29 (103 mg, 0.361 mmol, 91.1%) as a colorless syrup. Rf = 0.4 (CHCl3/MeOH/NH4OH (conc.), 1:2:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 12.4 (MeOH, c = 1.45); 1H NMR (300 MHz, D2O): δ = 3.80 (dd, 1H, J5,6a = 3.4, J6a,6b = 11.4 Hz, H-6a), 3.73–3.53 (m, 10H, H-3, H-6b, 2 × H-2′, 2 × H-3′, 2 × H-4′, 2 × H-5′), 3.22 (dd, 1H, J3,4 = J4,5 = 9.8 Hz, H-4), 3.10 (dd, 1H, H-2eq), 3.04 (dd, 1H, H-5aeq), 2.80 (t, 2H, J = 5.4 Hz, 2 × H-6′), 2.68 (t, 2H, J = 5.6 Hz, 2 × H-1′), 2.05 (dd, 1H, J5aax,5aeq = J5,5aax = 11.8 Hz, H-5aax), 2.04 (dd, 1H, J2ax,2eq = J2ax,3 = 10.7 Hz, H-2ax), 1.77 (m, 1H, H-5) ppm; 13C NMR (75.5 MHz, D2O): δ = 73.7 (C-4), 71.1 (C-3), 71.9, 69.5, 69.5, 67.7 (C-2′, C-3′, C-4′, C-5′), 60.7 (C-6), 57.6 (C-2), 54.6 (C-1′), 54.5 (C-5a), 42.8 (C-5), 39.8 (C-6′) ppm; MS (ESI-TOF): m/z calculated for [C12H26N2O5H] ([M + H]+) 279.1920, found 279.1927.
N-[2-[2-[2-[(3R,4R,5R)-3,4-Dihydroxy-5-(hydroxymethyl)-piperidin-1-yl]ethoxy]ethoxy]ethyl]-4-methylbenzenesulfonamide; or N-(8-(tosylamino)-3,6-dioxaoctyl)isofagomine (31, C19H32N2O7S)
Following general procedure C, compound 29 (52.3 mg, 0.19 mmol) was treated with 4-methylbenzene-1-sulfonyl chloride (42.9 mg, 0.23 mmol). Purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v) provided NHtosyl derivative 31 (61.0 mg, 0.14 mmol, 75.1%) as a colorless syrup. Rf = 0.5 (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 10.0 (MeOH, c = 0.90); 1H NMR (300 MHz, D2O): δ = 7.77 (d, 2H, J = 8.3 Hz, aromatic), 7.46 (d, 2H, J = 8.0 Hz, aromatic), 3.80 (dd, 1H, J5,6a = 3.4, J6a,6b = 11.4 Hz, H-6a), 3.70–3.46 (m, 10H, H-3, H-6b, 2 × H-2′, 2 × H-3′, 2 × H-4′, 2 × H-5′), 3.22 (dd, 1H, J3,4 = J4,5 = 9.8 Hz, H-4), 3.22 (t, 2H, J = 5.2 Hz, 2 × H-6′), 3.05 (m, 2H, H-2eq, H-5aeq), 2.66 (t, 2H, J = 5.6 Hz, 2 × H-1′), 2.43 (s, 3H, Ar–CH3 tosyl), 2.05 (dd, 1H, J5aax,5aeq = J5,5aax = 11.9 Hz, H-5aax), 2.04 (dd, 1H, J2ax,2eq = J2ax,3 = 10.5 Hz, H-2ax), 1.76 (m, 1H, H-5) ppm; 13C NMR (75.5 MHz, D2O): δ = 145.0, 135.3 (ipso), 130.1, 126.7 (aromatic), 73.7 (C-4), 71.1 (C-3), 69.4, 69.3, 68.8, 67.7 (C-2′, C-3′, C-4′, C-5′), 60.7 (C-6), 57.5 (C-2), 55.9 (C-1′), 54.5 (C-5a), 42.7 (C-5), 42.2 (C-6′), 20.7 (Ar–CH3 tosyl) ppm; MS (ESI-TOF): m/z calculated for [C19H32N2O7SH] ([M + H]+) 433.2008, found 433.2025.
N-[2-[2-[2-[(3R,4R,5R)-3,4-Dihydroxy-5-(hydroxymethyl)-piperidin-1-yl]ethoxy]ethoxy]ethyl]-5-(dimethylamino)-naphthalene-1-sulfonamide; or N-(8-(dansylamino)-3,6-dioxaoctyl)isofagomine (33, C24H37N3O7S)
Following general procedure C, compound 29 (43.1 mg, 0.15 mmol) was treated with 5-(dimethylamino)naphthalene-1-sulfonyl chloride (50.1 mg, 0.18 mmol). Subsequent purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 4/4/1 v/v/v) provided NH-dansylated derivative 33 (52.0 mg, 0.10 mmol, 65.6%) as a fluorescent wax. Rf = 0.4 (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 8.0 (MeOH, c = 0.88); 1H NMR (300 MHz, MeOD): δ = 8.49 (d, 1H, J = 8.5 Hz, aromatic), 8.28 (d, 1H, J = 8.6 Hz, aromatic), 8.14 (d, 1H, J = 7.3 Hz, aromatic), 7.52 (m, 2H, aromatic), 7.21 (d, 1H, J = 7.5 Hz, aromatic), 3.74 (dd, 1H, J5,6a = 3.7, J6a,6b = 11.0 Hz, H-6a), 3.54–3.21 (m, 10H, H-3, H-6b, 2 × H-2′, 2 × H-3′, 2 × H-4′, 2 × H-5′), 3.07 (m, 3H, H-2eq, H-4, H-5aeq), 2.99 (t, 2H, J = 6.8 Hz, 2 × H-6′), 2.82 (s, 6H, Ar–N(CH3)2 dansyl), 2.64 (t, 2H, J = 5.4 Hz, 2 × H-1′), 2.04 (dd, 1H, J5aax,5aeq = J5,5aax = 11.9 Hz, H-5aax), 2.01 (dd, 1H, J2ax,2eq = J2ax,3 = 11.0 Hz, H-2ax), 1.76 (m, 1H, H-5) ppm; 13C NMR (75.5 MHz, MeOD): δ = 153.2, 137.4, 131.2, 131.1 (ipso), 131.0, 130.0, 129.1, 124.3, 120.7, 116.4 (aromatic), 75.5 (C-4), 72.6 (C-3), 71.2, 71.1, 70.6, 69.0 (C-2′, C-3′, C-4′, C-5′), 62.4 (C-6), 59.5 (C-2), 58.2 (C-1’), 56.5 (C-5a), 45.8 (Ar–N(CH3)2 dansyl), 44.6 (C-5), 43.8 (C-6′) ppm; MS (ESI-TOF): m/z calculated for [C24H37N3O7SH] ([M + H]+) 512.2430, found 512.2454.
(3R,4R,5R)-1-(6-Azidohexyl)-5-(hydroxymethyl)piperidine-3,4-diol; or N-(6-azidohexyl)isofagomine (28, C12H24N4O3)
Following general procedure B, compound 1 (153 mg, 1.04 mmol) was treated with 1-azido-6-bromohexane (26, 236 mg, 1.14 mmol) [50]. Purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v) provided N-alkylated derivative 28 (182 mg, 0.67 mmol, 64.3%) as a pale yellow syrup. Rf = 0.4 (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 14.6 (MeOH, c = 0.96); 1H NMR (300 MHz, MeOD): δ = 3.82 (dd, 1H, J5,6a = 3.6, J6a,6b = 10.9 Hz, H-6a), 3.52 (m, 2H, H-3, H-6b), 3.30 (m, 2H, 2 × H-6′), 3.11 (dd, 1H, J3,4 = J4,5 = 9.8 Hz, H-4), 3.07–2.99 (m, 2H, H-2eq, H-5aeq), 2.39 (m, 2H, 2 × H-1′), 1.86 (dd, 1H, J2ax,2 eq = J2ax,3 = 11.0 Hz, H-2ax), 1.86 (dd, 1H, J5aax,5aeq = J5,5aax = 11.0 Hz, H-5aax), 1.79–1.70 (m, 1H, H-5), 1.67–1.49 (m, 4H, 2x H-2′, 2x H-5′), 1.48–1.30 (m, 4H, 2x H-3′, 2x H-4′) ppm; 13C NMR (75.5 MHz, MeOD): δ = 76.0 (C-4), 73.0 (C-3), 62.7 (C-6), 59.8 (C-2), 59.3 (C-1′), 56.5 (C-5a), 52.4 (C-6′), 44.9 (C-5), 29.8, 28.1, 27.7, 27.6 (C-2′, C-3′, C-4′, C-5′) ppm; MS (ESI-TOF): m/z calculated for [C12H24N4O3H] ([M + H]+) 273.1926, found 273.1936.
(3R,4R,5R)-1-(6-Aminohexyl)-5-(hydroxymethyl)piperidine-3,4-diol; or N-(6-aminohexyl)isofagomine (30, C12H26N2O3)
According to general procedure A, compound 28 (173 mg, 0.64 mmol) was converted into its corresponding crude amine 30. Subsequent purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v) provided compound 30 (133 mg, 0.54 mmol, 85.0%) as a colorless syrup. Rf = 0.2 (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 13.7 (MeOH, c = 1.03); 1H NMR (300 MHz, MeOD): δ = 3.81 (dd, 1H, J5,6a = 3.7, J6a,6b = 10.9 Hz, H-6a), 3.52 (m, 2H, H-3, H-6b), 3.13–2.98 (m, 3H, H-2eq, H-4, H-5aeq), 2.91 (t, 2H, J = 6.8 Hz, 2x H-6′), 2.43 (m, 2H, 2x H-1′), 1.90 (dd, 1H, J5aax,5aeq = J5,5aax = 11.1 Hz, H-5aax), 1.90 (dd, 1H, J2ax,2eq = J2ax,3 = 11.1 Hz, H-2ax), 1.79–1.49 (m, 5H, 2x H-2′, 2x H-5′, H-5), 1.48–1.30 (m, 4H, 2x H-3′, 2x H-4′) ppm; 13C NMR (75.5 MHz, MeOD): δ = 75.8 (C-4), 72.9 (C-3), 62.5 (C-6), 59.6 (C-2), 59.1 (C-1′), 56.4 (C-5a), 44.8 (C-5), 40.7 (C-6′), 28.5, 28.0, 27.4, 27.2 (C-2′, C-3′, C-4′, C-5′) ppm; MS (ESI-TOF): m/z calculated for [C12H26N2O3H] ([M + H]+) 247.2021, found 247.2024.
N-[6-[(3R,4R,5R)-3,4-Dihydroxy-5-(hydroxymethyl)piperidin-1-yl]hexyl]-4-methylbenzenesulfonamide; or N-((6-to-sylamino)hexyl)isofagomine (32, C19H32N2O5S)
Following general procedure C, compound 30 (45.0 mg, 0.18 mmol) was treated with 4-methylbenzene-1-sulfonyl chloride (41.8 mg, 0.22 mmol). Purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v) provided NHtosyl derivative 32 (52.0 mg, 0.1323 mmol, 71.1%) as a colorless syrup. Rf = 0.4 (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v); \({\left[\alpha \right]}_{D}^{20}\)= 14.5 (MeOH, c = 1.10); 1H NMR (300 MHz, MeOD): δ = 7.73 (d, J = 8.3 Hz, 2H, aromatic), 7.38 (d, J = 8.0 Hz, 2H, aromatic), 3.83 (dd, 1H, J5,6a = 3.7, J6a,6b = 11.0 Hz, H-6a), 3.52 (m, 2H, H-3, H-6b), 3.10 (dd, 1H, J3,4 = J4,5 = 9.2 Hz, H-4), 3.07–2.99 (m, 2H, H-2eq, H-5aeq), 2.83 (t, 2H, J = 6.8 Hz, 2 × H-6′), 2.44 (s, 3H, Ar–CH3 tosyl), 2.42–2.33 (m, 2H, 2 × H-1′), 1.90 (dd, 1H, J5aax,5aeq = J5,5aax = 11.0 Hz, H-5aax), 1.89 (dd, 1H, J2ax,2 eq = J2ax,3 = 11.0 Hz, H-2ax), 1.75 (m, 1H, H-5), 1.57–1.38 (m, 4H, 2 × H-2′, 2 × H-5′), 1.37–1.19 (m, 4H, 2 × H-3′, 2 × H-4′) ppm; 13C NMR (75.5 MHz, MeOD): δ = 144.5, 139.1 (ipso), 130.7, 128.0 (aromatic), 75.9 (C-4), 73.0 (C-3), 62.6 (C-6), 59.6 (C-2), 59.2 (C-1′), 56.4 (C-5a), 44.9 (C-5), 43.9 (C-6′), 30.5, 28.0, 27.5, 27.5 (C-2′, C-3′, C-4′, C-5′), 21.4 (Ar–CH3 tosyl) ppm; MS (ESI-TOF): m/z calculated for [C19H32N2O5SH] ([M + H]+) 401.2110, found 401.2124.
4-Bromo-N-[6-[(3R,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)-piperidin-1-yl]hexyl]benzenesulfonamide; or N-((6-brosylamino)hexyl)isofagomine (34, C18H29BrN2O5S)
Following general procedure C, compound 30 (42.0 mg, 0.17 mmol) was treated with 4-bromobenzene-1-sulfonyl chloride (52.3 mg, 0.2046 mmol). Purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v) provided NHbrosyl derivative 34 (57.1 mg, 0.12 mmol, 71.8%) as a white solid. Rf = 0.4 (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v); \({\left[\alpha \right]}_{{\text{D}}}^{20}\)= + 8.0 (MeOH, c = 1.00); 1H NMR (300 MHz, MeOD): δ = 7.75 (s, 4H, aromatic), 3.83 (dd, 1H, J5,6a = 3.7, J6a,6b = 10.9 Hz, H-6a), 3.54 (m, 2H, H-3, H-6b), 3.26–3.03 (m, 3H, H-2 eq, H-4, H-5aeq), 2.86 (t, J = 6.8 Hz, 2H, 2x H-6′), 2.44 (m, 2H, 2x H-1′), 1.98 (dd, 1H, J5aax,5aeq = J5,5aax = 11.7 Hz, H-5aax), 1.96 (dd, 1H, J2ax,2 eq = J2ax,3 = 11.0 Hz, H-2ax), 1.75 (m, 1H, H-5), 1.56–1.38 (m, 4H, 2x H-2′, 2 × H-5′), 1.34–1.24 (m, 4H, 2x H-3′, 2x H-4′) ppm; 13C NMR (75.5 MHz, MeOD): δ = 141.4, 128.0 (ipso), 133.4, 129.8 (aromatic), 75.6 (C-4), 72.7 (C-3), 62.4 (C-6), 59.3 (C-2), 59.1 (C-1′), 56.2 (C-5a), 44.7 (C-5), 43.9 (C-6′), 30.5, 27.9, 27.4, 27.3 (C-2′, C-3′, C-4′, C-5′) ppm; MS (ESI-TOF): m/z calculated for [C18H29BrN2O5SH] ([M + H]+) 467.1038, found 467.1043.
N-[6-[(3R,4R,5R)-3,4-Dihydroxy-5-(hydroxymethyl)piperidin-1-yl]hexyl]-5-(dimethylamino)naphthalene-1-sulfonamide; or N-((6-dansylamino)hexyl)isofagomine (35, C24H37N3O5S)
Following general procedure C, compound 30 (43 mg, 0.17 mmol) was treated with 5-(dimethylamino)naphthalene-1-sulfonyl chloride (56.5 mg, 0.2095 mmol). Purification on silica gel (CHCl3/MeOH/NH4OH (conc.), 4:4:1 v/v/v) provided NH-dansylated derivative 35 (49.2 mg, 0.10 mmol, 58.5%) as a fluorescent wax. Rf = 0.3 (CHCl3/MeOH/NH4OH (conc.), 8:4:1 v/v/v); \({\left[\alpha \right]}_{D}^{20}\)= + 9.8 (MeOH, c = 1.22); 1H NMR (300 MHz, MeOD): δ = 8.56 (d, 1H, J = 8.5 Hz, aromatic), 8.36 (d, 1H, J = 8.5 Hz, aromatic), 8.19 (d, 1H, J = 7.5 Hz, aromatic), 7.62–7.53 (m, 2H, aromatic), 7.28 (d, 1H, J = 7.5 Hz, aromatic), 3.82 (dd, 1H, J5,6a = 3.5, J6a,6b = 10.9 Hz, H-6a), 3.51 (m, 2H, H-3, H-6b), 3.08 (dd, 1H, J3,4 = J4,5 = 9.3 Hz, H-4), 2.97 (m, 2H, H-2eq, H-5aeq), 2.89 (s, 6H, Ar–N(CH3)2 dansyl), 2.84 (m, 2H, 2 × H-6′), 2.24 (m, 2H, 2 × H-1′), 1.82 (dd, 1H, J5aax,5aeq = J5,5aax = 11.2 Hz, H-5aax), 1.80 (dd, 1H, J2ax,2eq = J2ax,3 = 11.2 Hz, H-2ax), 1.30 (m, 1H, H-5), 1.34–1.24 (m, 4H, 2x H-2′, 2 × H-5′), 1.19–0.98 (m, 4H, 2x H-3′, 2x H-4′) ppm; 13C NMR (75.5 MHz, MeOD): δ = 153.2, 137.3, 131.2, 131.0 (ipso), 131.1, 130.2, 129.1, 124.3, 120.7, 116.4, (aromatic), 76.1 (C-4), 73.0 (C-3), 62.7 (C-6), 59.7 (C-2), 59.2 (C-1′), 56.5 (C-5a), 45.9 (Ar–N(CH3)2 dansyl), 44.9 (C-5), 43.8 (C-6′), 30.4, 28.0, 27.5, 27.3 (C-2′, C-3′, C-4′, C-5′) ppm; MS (MALDI-TOF): m/z calculated for [C24H37N3O5SNa] ([M + Na]+) 502.2352, found 502.2362.
Kinetic studies with compounds 1 and 27–35
Enzyme activities were assayed spectrophotometrically using 96-well plates from SARSTEDT on a Spark® Multimode Microplate Reader (TECAN Group AG, Switzerland). In the inhibition kinetic experiments, three different substrate concentrations ([S] < Km, [S] = Km, [S] > Km) were used. For each substrate concentration, the before optimized concentration of enzyme was pre-incubated with the inhibitors at seven different concentrations and one control sample without inhibitor, referring to 100% of activity at the specific conditions. The initial rate was determined by measuring the release of p-nitrophenol at the expense of p-nitrophenyl glycoside specific for each enzyme at 405 nm for up to 5 min. Initial rates were calculated in Excel and Lineweaver–Burk plots (1/v vs. 1/[S] for each inhibitor concentration) were constructed to validate the use of competitive, non-competitive or mixed-type inhibition models and to assess the fit of the data. The data were then fit to the appropriate inhibition model using non-linear regression analysis with GraFit 7.0.3. (Erithacus Software).
Specific assay conditions for each enzyme:
Agrobacterium sp. β-glucosidase GH1 (Abg): 50 mM sodium phosphate buffer (pH 7) at 25 °C. Substrate: pNP β-Glc, Km = 0.2 mM, [S] = 0.1 mM, 0.2 mM, 0.4 mM.
Thermotoga maritima β-glucosidase GH1 (T. mar.): 50 mM sodium maleate buffer (pH 6.5) at 37 °C. Substrate: pNP β-Glc, Km = 0.7 mM, [S] = 0.4 mM, 0.7 mM, 1.4 mM [56].
Prunus dulcis β-glucosidase GH1 (P. dul., sweet almonds): 50 mM sodium phosphate buffer (pH 7) at 25 °C. Substrate: pNP β-Glc, Km = 4.8 mM, [S] = 3.6 mM, 4.8 mM, 6.0 mM [56].
Saccharomyces cerevisiae α-glucosidase GH13 (S. cer.): 50 mM sodium phosphate buffer (pH 7) at 25 °C. Substrate: pNP α-Glc, Km = 3.2 mM, [S] = 1.6 mM, 3.2 mM, 6.3 mM.
Escherichia coli β-galactosidase GH2 (E.coli): 50 mM sodium phosphate buffer, 1.0 mM MgCl2 (pH 7) at 25 °C. Substrate: pNP β-Gal, Km = 53 µM, [S] = 35 µM, 55 µM, 100 µM.
Canavalia ensiformis α-mannosidase GH38 (C. ens.): 50 mM sodium phosphate buffer (pH 7) at 25 °C. Substrate: pNP α-Manno, Km = 0.9 mM, [S] = 0.5 mM, 0.9 mM, 2.0 mM.
Data availability
Not applicable.
References
Jespersen TM, Dong W, Sierks MR, Skrydstrup T, Lundt I, Bols M (1994) Angew Chem Int Ed 33:1778
Wennekes T, van den Berg RJBHN, Boot RG, van der Marel GA, Overkleeft HS, Aerts JMFG (2009) Angew Chem 121:9006
Meloncelli PJ, Stick RV (2006) Aust J Chem 59:827
Yu Z, Sawkar AR, Whalen LJ, Wong C-H, Kelly JW (2007) J Med Chem 50:94
Malik G, Guinchard X, Crich D (2012) Org Lett 14:596
Oikonomakos NG, Tiraidis C, Leonidas DD, Zographos SE, Kristiansen M, Jessen CU, Nørskov-Lauritsen L, Agius L (2006) J Med Chem 49:5687
Jakobsen P, Lundbeck JM, Kristiansen M, Breinholt J, Demuth H, Pawlas J, Candela MP, Andersen B, Westergaard N, Lundgren K, Asano N (2001) Bioorg Med Chem 9:733
Wang B, Bogh SA, Poulsen JCN, Laursen BW, Bols M (2020) Eur J Org Chem 2020:3989
Lindbäck E, Laursen BW, Poulsen JCN, Kilså K, Pedersen CM, Bols M (2015) Org Biomol Chem 13:6562
Malik G, Ferry A, Guinchard X, Cresteil T, Crich D (2013) Chem Eur J 19:2168
Greimel P, Häusler H, Lundt I, Rupitz K, Stütz AE, Tarling CA, Withers SG, Wrodnigg TM (2006) Bioorg Med Chem Lett 16:2067
Ichikawa Y, Igarashi Y, Ichikawa M, Suhara Y (1998) J Am Chem Soc 120:3007
Zhu X, Sheth KA, Li S, Chang H-H, Fan J-Q (2005) Angew Chem Int Ed 44:7450
Moréra S, Vigouroux A, Stubbs KA (2011) Org Biomol Chem 9:5945
Varrot A, Tarling CA, Macdonald JM, Stick RV, Zechel DL, Withers SG, Davies GJ (2003) J Am Chem Soc 125:7496
Williams RJ, Iglesias-Fernández J, Stepper J, Jackson A, Thompson AJ, Lowe EC, White JM, Gilbert HJ, Rovira C, Davies GJ, Williams SJ (2014) Angew Chem Int Ed 53:1087
Hakki Z, Thompson AJ, Bellmaine S, Speciale G, Davies GJ, Williams SJ (2015) Chem Eur J 21:1966
Schuster M (1999) Bioorg Med Chem Lett 9:615
Allen KA, Brown RL, Norris G, Tyler PC, Watt DK, Zubkova OV (2010) Carbohydr Res 345:1831
van den Berg RJBHN, Donker-Koopman W, van Boom JH, Aerts HMFG, Noort D (2004) Bioorg Med Chem 12:891
Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, Rose C, Billette de Villemeur T, Berger MG (2017) Int J Mol Sci 18:441
Lieberman RL, Wustman BA, Huertas P, Powe AC, Pine CW, Khanna R, Schlossmacher MG, Ringe D, Petsko GA (2007) Nat Chem Biol 3:101
Brumshtein B, Greenblatt HM, Butters TD, Shaaltiel Y, Aviezer D, Silman I, Futerman AH, Sussman JL (2007) J Biol Chem 282:29052
Zechel DL, Boraston AB, Gloster T, Boraston CM, Macdonald JM, Tilbrook DMG, Stick RV, Davies GJ (2003) J Am Chem Soc 125:14313
Dax K, Gaigg B, Grassberger V, Kölblinger B, Stütz AE (1990) J Carbohydr Chem 9:479
Trnka T, Černý M (1971) Collect Czech Chem Commun 36:2216
Andersch J, Bols M (2001) Chem Eur J 7:3744
Best WM, Macdonald JM, Skelton BW, Stick RV, Tilbrook DMG, White AH (2002) Can J Chem 80:857
Goddard-Borger ED, Stick RV (2007) Aust J Chem 60:211
Panfil I, Solecka J, Chmielewski M (2006) J Carbohydr Chem 25:673
Spanu P, Candia CD, Ulgheri F (2010) Tetrahedron Lett 51:2400
Espeel P, Piens K, Callewaert N, van der Eycken J (2008) Synlett 2008:2321
Imahori T, Ojima H, Yoshimura Y, Takahata H (2008) Chemistry 14:10762
Imahori T, Ojima H, Tateyama H, Mihara Y, Takahata H (2008) Tetrahedron Lett 49:265
Guanti G, Riva R (2003) Tetrahedron Lett 44:357
Banfi L, Guanti G, Paravidino M, Riva R (2005) Org Biomol Chem 3:1729
Ouchi H, Mihara Y, Watanabe H, Takahata H (2004) Tetrahedron Lett 45:7053
Pandey G, Kapur M (2000) Tetrahedron Lett 41:8821
Pandey G, Kapur M (2001) Synthesis 112:1263
Hansen SU, Bols M (2000) J Chem Soc Perkin Trans 1:911
Takahata H, Mihara Y, Ojima H, Imahori T, Yoshimura Y, Ouchi H (2007) Heterocycles 72:633
Altenbach H-J, Blanda G (1998) Tetrahedron Asymmetry 9:1519
Zhao G, Deo UC, Ganem B (2001) Org Lett 3:201
Yoshikawa M, Okaichi Y, Cheon Cha B, Kitagawa I (1990) Tetrahedron 46:7459
Singh N, Pandey J (2020) Mini-Rev Org Chem 17:297
Rosenthal A, Schöllnhammer G (1972) Can J Chem 50:1780
Berliner MA, Belecki K (2005) J Org Chem 70:9618
Roy BC, Santos M, Mallik S, Campiglia AD (2003) J Org Chem 68:3999
Shon Y-S, Kelly KF, Halas NJ, Lee TR (1999) Langmuir 15:5329
Steiner AJ, Stütz AE, Tarling CA, Withers SG, Wrodnigg TM (2007) Carbohydr Res 342:1850
Weber P, Thonhofer M, Averill S, Davies GJ, Santana AG, Müller P, Nasseri SA, Offen WA, Pabst BM, Paschke E, Schalli M, Torvisco A, Tschernutter M, Tysoe C, Windischhofer W, Withers SG, Wolfsgruber A, Wrodnigg TM, Stütz AE (2020) Molecules 25:4025
Thonhofer M, Weber P, Santana AG, Fischer R, Pabst BM, Paschke E, Schalli M, Stütz AE, Tschernutter M, Windischhofer W, Withers SG (2016) Bioorg Med Chem Lett 26:1438
Thonhofer M, Weber P, Gonzalez Santana A, Tysoe C, Fischer R, Pabst BM, Paschke E, Schalli M, Stütz AE, Tschernutter M, Windischhofer W, Withers SG (2016) Carbohydr Res 429:71
Lundt I, Steiner AJ, Stütz AE, Tarling CA, Ully S, Withers SG, Wrodnigg TM (2006) Bioorg Med Chem 14:1737
Prasch H, Wolfsgruber A, Thonhofer M, Culum A, Mandl C, Weber P, Zündel M, Nasseri SA, Gonzalez Santana A, Tegl G, Nidetzky B, Gruber K, Stütz AE, Withers SG, Wrodnigg TM (2023) ChemBioChem 24:e202300480
Keyzor I, Shohet S, Castelli J, Sitaraman S, Veleva-Rotse B, Weimer JM, Fox B, Willer T, Tuske S, Crathorne L (2023) Biomolecules 13:1227
Acknowledgements
We are grateful for NOE-experiment of compound 22 to Ao.Univ.-Prof. Dipl.-Ing. Dr.techn. Hans-Jörg Weber. We thank Dipl.-Ing. Dr.rer.nat. BSc Markus Hochegger-Krawanja and Markus Köck for conducted MS analysis of compounds 20–23 and 27–34.
Funding
Open access funding provided by Graz University of Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Culum, A., Prasch, H., Dorn, T. et al. A remarkable change in inhibition potency and selectivity of isofagomine by simple N-modification. Monatsh Chem (2024). https://doi.org/10.1007/s00706-024-03210-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00706-024-03210-7