
Abstract
Online electrochemical mass spectrometry (OEMS) is a promising analytical technique to monitor minor side reactions with gaseous species, taking place while charging and discharging a lithium-ion cell. However, besides the manifold examples of these custom-made systems and their application, a clear analytical view on the origin of the evolving gasses and their manifold interactions within the cell environment is missing and therefore given in this work. To get a better understanding of the complexity of gas evolution associated with electrochemical reactions in lithium-ion cells, the use of chronoamperometry as an analytical method was chosen. This led to a precise variation of the applied voltage and voltage-pulse length and enabled therewith a clear starting point of the electrochemically triggered reactions. It was found that chronoamperometry can be used to precisely trigger those reactions with gaseous products. Additionally, it was found that the release of gaseous species depends on many parameters including the cell configuration, the current, and the gas species. The response time determination showed that a custom-made highly porous electrode configuration had an overall better response behavior within minutes with differences for the respective gasses of interest, compared to a standard foil configuration cells. The herein presented methodology shows how an electroanalytical approach can help gain further insight into advanced hyphenated methods, such as OEMS in the context of studies of lithium-ion cells.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To fulfill the ambitious CO2 targets within the Paris climate protocol, the need for lithium-ion cells (LICs) has grown exponentially within recent years. Mainly the demand for automotive and further public transportation purposes, such as busses and ferries, leads to a growing amount of cell factories around the globe [1].
However, to use the full potential of future LICs, their ageing must be understood in more detail [2, 3]. One growing aspect is the gas evolution during the very first charge. Hereby, the liquid electrolyte is partially decomposed into hydrogen (H2), ethylene (C2H4), carbon monoxide (CO), and carbon dioxide (CO2), while forming the solid electrolyte interface (SEI) on the negative electrode [4,5,6,7]. To track these gaseous byproducts, advanced analytical techniques with high selectivity and sensitivity are needed. A sophisticated approach is made by the combination of electrochemical cycling procedures with mass spectrometry (MS) [8,9,10,11].
Imhof and Novak [12,13,14], as well as McCloskey et al. [15] pioneered the use differential electrochemical mass spectrometry (DEMS) to observe the gas evolution during SEI formation. Their setup allowed a discrete sampling of the gas species from a semi-closed cell headspace probed by MS. Alternatively, Gasteiger et al. [16] developed an online electrochemical mass spectrometry (OEMS) system. A closed cell headspace is directly connected to the MS with a crimped stainless steel capillary. However, despite all these custom-made setups and their proof-of-concept studies, a clear and critical analytical description of the overall interactions of the complex processes taking place in a LIC and their influence onto the response behavior is still missing. In the context of this work, the response time (tR) is defined as the time between the electrochemical event (EC Event) and the recording of the gas species in the MS, see Fig. 1.

Schematic representation of the response time description of an OEMS system
In this report, the electrochemical response behavior of a model cell for the usage of electrochemically coupled mass spectrometry is described. Chronoamperometry (CA) as a simple electroanalytical method was used to generate a well-defined stimulus for a short time interval to follow the response characteristics of the respective experimental configuration. The two parameters, namely the applied potential and the pulse duration, were used to precisely trigger the electrochemical reactions, when charging a cell for the first time. The simultaneously recorded mass spectra were used to evaluate the responding gassing behavior of the cell. The study was carried out with two different electrode cell configurations. First, a model system with standard foil coating was used. Then, a more detailed investigation was carried out using a highly porous and gas permeable mesh coating.
Results and Discussion
1) Challenges in online gas analysis for LICs
In this chapter, the most important parameters, which have an influence on the response behavior and therefore onto the definition of online gas analysis are described. The response time of a LIC to the applied electrochemical stimulus in terms of gas generation will depend mainly on the setup itself and on the cell configuration. Usually, LIC electrodes are coated on either copper or aluminum foil, which are impermeable for the evolving gasses. An alternative to this configuration is a gas permeable electrode, where the active material is coated on a mesh substrate. See the experimental section and Fig. 6 for a detailed illustration of the two configurations used in this study. The mesh provides a regular spacing in the order of micrometers, which results in a highly porous and gas permeable electrode. Using standard foil coated electrodes, the formation of gas bubbles and a possible bubble entrapment in the porous active material can be assumed. The microscopic bubble formation will depend on the quantity of formed gasses (massive vs. small gassing). Hence, it is a function of the interval of the passing current, the surface area of the active material, the gas species as well as the overall cell chemistry.
After formation, a partitioning of the gasses between the active solid material, the liquid electrolyte phase, and the gas phase will take place. Each of these phases allows several degrees of freedom for interactions. Therefore, the individual properties for each gas, in each phase must be considered on its own. Another important parameter is the reactivity of the produced gasses within the electrochemical surrounding of a LIC. The gasses have also different solubility properties in the liquid phase [17, 18]. Especially, carbon dioxide shows an increased solubility in common electrolyte components, such as ethylene carbonate. Due to their higher solubility, evolving CO2 molecules tend more likely to dissolve in the liquid phase and delays in the MS detection might occur. Further important properties are the molecule size, pressure, temperature, conductivity, and viscosity.
In addition, the interaction between the gasses and all other LIC components has to be considered. In this context, the liquid electrolyte plays a crucial role. A solution of lithium hexafluorophosphate in organic carbonate blends represents the state-of-the-art electrolyte. However, especially the conducting salt, e.g., LiPF6 shows a strong sensitivity towards water impurities and forms reactive by-products, e.g., hydrofluoric acid (HF). These compounds may react with the evolving gasses or the organic carbonates, to produce even those gasses of interest, e.g., hydrogen or carbon monoxide. Some carbonates meanwhile have a high vapour pressure and produce MS signals with high intensity, which may overlay with the signals from the gasses of interest [19]. To reduce this influence, the LIC is usually hold at open circuit conditions (OCV) for several hours to obtain a stable background signal for each mass channel. In case of high vapor pressures or complex electrolyte mixtures, the background signal does not converge, and a more complex calculation routine (exponential fitting routine) has to be used to differentiate between electrochemically produced gasses and the ongoing increase of degradation-related gasses.
After their formation and manifold interactions within the material mix, the formed gasses diffuse into the cell headspace. In a mesh configuration, the diffusion length is in order of micrometers, whereas for a foil configuration, the distances are much longer (order of millimeters). The retention of the gasses in this configuration will strongly depend on their exact place of origin within the electrode assembly. The gasses which are formed on the outer side, can diffuse fast into the cell headspace, whereas gasses formed in the center of the electrode diffuse through the electrode until they reach the outer edge of the electrode. The diffusion for each species is mainly a function of solubility, molecule size, and temperature, and can be expressed by the diffusion coefficient of each gas species.
The cell headspace itself provides a diffusion length in the order of centimeters. However, the diffusion in the gas phase is comparatively fast. The molecules can than easily enter the ultra-high vacuum (UHV) regime where they are recorded by the MS. In our case, the barrier between cell headspace and UHV regime is made of a crimped capillary, where a porous metal filter allows a one-stage pressure reduction [20]. This flow restrictor provides a uniform gas flow with minimal friction. However, the dwell time within this restrictor strongly depends on the viscosity of the gas and the size of the gas molecules.
All herein described phenomena provide only a short summary about the most obvious factors, which must be considered, when it comes to online gas analysis within LICs. All these factors strongly interfere with each other and are not taking place separately. Therefore, a clear definition or separation of these processes is challenging. While using typical cycling protocols, e.g., galvanostatic cycling with potential limitation, it also has to be considered, that the hereby monitored current is associated with a higher transient response, where even the starting point of the gas evolution can’t be clearly defined. Rough estimation only based on the diffusion length implies that the evolving gasses can be detected within minutes.
To answer the analytical question of the response behavior between the electrochemically triggered gas evolution and the corresponding detection in the MS, a clearly defined starting point of the electrochemical reaction itself, is needed first. This is usually not the case, when cycling LICs with standardized, e.g., galvanostatic methods. The use of CA allows to instantly step the potential of the working electrode to a well-defined potential and therefore having a clearly defined starting point (t0) of the electrochemical reaction. Furthermore, CA allows a precise variation of the applied potential and pulse length of the applied potential.
2) Study of the influence of the applied potential
At first, investigations concerning the influence of the applied potential regime were carried out by using the mesh configuration LIC only. A starting potential of 2.0 V and an upper cut-off voltage of 4.5 V were chosen as the potential window of interest. The potential was held for four seconds in each experiment, whereby the potential was stepwise increased by 0.5 V for each measurement. The applied CA protocol and the resulting current data are depicted on the left side of Fig. 2. The simultaneously recorded mass spectra, for the individual gas species are depicted on the right side of Fig. 2. The vertical dashed lines indicate the times for the application of short potential pulses for the CA measurements. As it is out of the scope of this manuscript to exactly describe the different chemical processes which occur in a LIC, only the general behavior for each configuration will be discussed briefly.

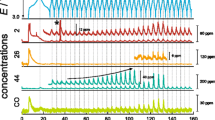
Left: Applied potential (top, blue) and recorded current (bottom, green) during the applied voltage variation under chronoamperometric conditions with a pulse length of four seconds each. Right: Corresponding gassing-response behavior, recorded by the mass spectrometer for the gasses of interest. For better comparison, the intensities were normalized to the 36Ar isotope (denoted as Ix/I36Ar) (color figure online)
For the use of the lowest potential of 2.0 V, already a slight increase in the CO2 (purple) signal can be seen. In this potential region, the decomposition of various impurities is expected [21, 22]. The increase of the applied potential to 2.5 V and 3.0 V results in a further sharp and significant rise of the CO2 mass trace. This indicates that the amount of released gasses strongly depends on the passing current. The use of the nominal potential of 4.0 V reveals the expected gassing behavior during the first charge of the cell. Note, that an expected gas evolution is determined as the point, where a response of all here shown mass traces can be seen. The application of a potential of 4.5 V would normally lead to a decomposition of most cell materials. But as these processes are relatively slow and therefore, the applied potential sequence was probably too short to result in that kind of decomposition as this would result in a higher amount of CO2 evolution [23]. The MS signals for the gasses ethylene (red) and carbon monoxide (orange) already start to increase, using lower potentials. It is most likely that some electrochemical reactions are already been triggered earlier. For example, it is known that impurities, like water or HF, trigger the decomposition of the electrolyte in an early stage if the cell is only assembled and not charged, as only the SEI protects the cell components from further degradation [21, 22]. These degradation processes most likely end up, by producing ethylene and carbon monoxide. No obvious change in the hydrogen (blue mass trace) can be seen. The stepwise decrease in the oxygen mass trace (green) is most probably the result of a minor amount of oxygen, which is reduced shortly after the potential is applied. However, this series shows clearly how the CA approach can be used to trigger specific electrochemical reactions and to monitor their consequences on the gassing behavior. It is important to note that even in the context of a simple CA experiment, differences concerning the response behavior for the formation, release, and detection of various gasses can be seen.
3) Investigation of the influence of the pulse length of CA experiments
As second part of this study, the variation of the pulse length was investigated. In this chapter both cell configurations, namely the mesh configuration as well as the foil configuration, were used to investigate how the pulse length effects the overall gassing behavior for different configurations. According to the former experiments, where a significant gassing behavior was observed for potential pulses between 4.0 and 4.5 V, a cut-off potential of 4.2 V was used in this study. The potential pulse durations and the recorded chronoamperometric currents are depicted in Fig. 3.

Applied potential pulses (top, blue) of varying durations and recorded currents (bottom, green) for chronoamperometric measurements using a foil configuration LIC (left) and a mesh configuration LIC (right) (color figure online)
The pulse length ranged from one second up to four seconds for the mesh configuration and up to seven seconds for the foil configuration. The latter parameter setting was applied in order to generate a similar amount of gas (signal intensities) as for the mesh configuration. The recorded currents fit to the earlier performed potential variation series. Here, the potential variation was also performed for the mesh configuration and showed a similar current behavior in the potential regime between 4.0 and 4.5 V. The simultaneously recorded mass spectra, for the individual gas species are depicted in Fig. 4. For the foil configuration (left), no sufficient gas evolution can be assigned to the CA pulse until a pulse length of five seconds is reached.

Detected gas species during pulse length variation for either foil configuration LIC (left), or mesh configuration LIC (right). Note the different y-axis scale
However, an apparently linear increase of the intensities assigned to CO and C2H4 can be seen for shorter pulse lengths and might be attributed to early formed reactive by-products [2]. In case of the mesh configuration LIC (right part of Fig. 4), a shorter pulse length of three seconds is already sufficient to produce the expected gaseous species in a significant amount. Even the shortest applied pulse length of one second reveals a direct influence on the CO2 intensity. The CO2 behavior is special in several aspects. It is the only mass trace which shows an increasing as well as a decreasing behavior, directly related to the performed CA experiment. This behavior points out that besides gas evolution, the assumed gas consumption or dissolution processes must also be considered. However, besides the CO2 behavior, the linear increase of ethylene and carbon monoxide can be seen here. This suggests the assumption that ethylene is already been formed at shorter pulse sequences.
By comparing both configurations, it can be concluded that the overall response behavior for each configuration differs from each other. For the foil configuration, a longer pulse length is needed to produce the same amount of gas as in the case of the mesh configuration. For the latter configuration, the response behavior is more pronounced, meaning that higher sensitivity (amount of gasses formed) and a faster response is achieved. Consequently, the pulse length can be kept rather short. The gassing behavior in the foil configuration can be distinguished into two processes. First, an abrupt increase of the MS signals due to the CA stimulus can be seen for all mass traces, which is similar as for the mesh configuration, but much less in terms of absolute intensities. Then, the signal increase lowers its slope and changes to a more linear function. One explanation for this behavior could be that under these circumstances, the produced gasses are entrapped in the electrode assembly. The gasses, which are formed on the outer side of the electrode, can access the gaseous headspace faster as their diffusion distance is rather short. This results in the abrupt increase of the MS signals. Gasses which are generated close to the center of the electrode must diffuse through the laterally aligned electrode stack until they reach the outer part of the electrode. This results in a linear increase of the MS signals. For the mesh configuration, the produced gasses can easily diffuse through the highly porous material and are detected by the MS, as most active material seems to be accessible to the headspace. Consequently, the mesh configuration provides more pronounced signal intensities and reveal even fast changes in the gaseous headspace composition.
4) Evaluation of the response time characteristics
Finally, a precise evaluation of the response time was carried out. Thus, a detailed view of the individual mass traces in a rather short time interval after the application of the potential pulse is useful. Due to its more pronounced response characteristics regarding the CA stimulus and the MS signal increase, the mesh configuration, with a pulse sequence up to 4.2 V and four seconds, was chosen (see left part of Fig. 5).

Left: Detailed view of MS intensities of various gasses after one CA experiment, using a pulse length of four seconds and a potential of 4.2 V, using a mesh configuration LIC. Right: Relative mass signal intensities to determine the response time for either 63.2% or 99.3% (dashed lines)
To describe the response time, the following procedure was performed. First, the initial intensities, before the CA procedure, were read out (I0). The intensities after 20 min (I20) were used as final value (100%). The response time was determined analog to time constant definition typically used in the context of sensor characterization [24]. Two values for each mass channel were determined. The first value corresponds to 63.2% and the second to 99.3% of the final intensities.
The relative signal intensity for the mass channels of interest are plotted in Fig. 5. All response time data are furthermore depicted in Table 1. The focus was first set on the 63.2% benchmark. The fastest response is achieved for hydrogen, followed by oxygen, and carbon dioxide. Ethylene and carbon monoxide show the slowest response characteristics. This behavior can be explained with the origin of the evolved gas species. It is necessary to differentiate between directly consumed (oxygen) and produced gasses (hydrogen) or the gasses which are formed due to the SEI formation. These gas species are only the by-product of a cascade of several electrochemically triggered reactions at a certain potential inside the LIC. So, the origin and the response behavior of those produced gasses (ethylene, carbon monoxide, and carbon dioxide) are different from those of the gasses hydrogen and oxygen. However, all gasses reach the benchmark within seven minutes, after the CA experiment was performed.
The response time to achieve 99.3% of the final value is much longer for all gasses, taking up to 22 min. Hydrogen shows a special behavior. It has the fastest response time to achieve 63.2% of the total signal intensity but takes the longest time to achieve the 99.3% signal intensity. This indicates that besides gas evolution also, gas consumption is a part which has to be considered. Hydrogen is quickly formed but can be consumed fast within the cell environment. Furthermore, it has to be considered that hydrogen is challenging to measure within the MS due to its permeative properties. The results discussed above show that from an analytical point of view, the response time for each gas can be differentiated. However, the reason for the differences in response characteristics depends on many intrinsic parameters, and on the cell configuration itself. In addition, the cell chemistry has an important influence. Chemical processes, like the SEI formation are relatively slow processes. As the SEI is a fundamental key factor for the overall cell lifetime as well as for the performance of the cell, its correct formation is a crucial and cost-intensive part of every LIC manufacturing process. The usually applied very first cycling protocol is most likely in the range of hours up to days to form an ideal and homogeneous layer of this slowly formed SEI. Therefore, the presented response time characterization can be summarized toward a general response time which is for both cases in the range of several minutes. This estimation is strongly influenced by the amount of the released gasses. A further aspect is that under the described experimental conditions, the amount of gas evolution was sometimes close to the lower detection limit of the overall setup. When cycling a cell, a much higher gas release is expected, which will further affect the response time toward a faster response behavior.
Conclusion
In this work, the response characteristics of a custom-made online electrochemical mass spectrometry system for gas analysis in lithium-ion cells are presented. The results were achieved using CA to trigger electrochemical reactions within a cell under the release of gaseous compounds. As the gassing is a very complex interaction of gas evolution as well as gas consumption within a manifold environment, e.g., solid active material and liquid electrolyte components, a variety of parameters, such as diffusion, solubility, viscosity, and reactivity of the individual gas species, play an important role when it comes to online measurement techniques of those gasses.
CA was used as it provided a clear starting point of the electrochemical reaction itself to reduce the uncertainty in comparison to typically used cycling protocols. The advantage of short voltage pulses during the CA experiment was that all formed gaseous species can clearly be assigned to the rather short interval of the applied electrochemical stimulus.
While investigating first the potential range from 2.0 to 4.5 V and the corresponding effect on the gassing behavior, it was found that the CA technique can be used to trigger specific electrochemical reactions, e.g., water splitting and to investigate the dependency on the applied potential toward the individual underlying chemical reactions.
In addition, the effect of the pulse length of the potential jump on the gassing behavior was investigated for two typical LIC configurations. It was found that the adapted highly porous mesh configuration LIC provides much faster response characteristics toward the applied potential pulse. The foil configuration LIC showed an overall similar behavior, but much less pronounced. Furthermore, it was found that in case of the foil configuration LIC, delays of the transport of the formed gasses occurred by trapping them inside the foil electrode assembly. However, the foil configuration LIC is more representative for the typical LIC situation. A more detailed investigation of the overall response characteristics for a mesh configuration LIC revealed the complexity and challenge of pointing out a defined response time for the overall system.
This study demonstrated the intricate nature of online gas analysis of LICs using a simple electrochemical stimulus through a chronoamperometry (CA) experiment. The relevant gasses were detected within minutes, although with individual variations in response characteristics. The findings imply that the interpretation of more complex LIC experiments conducted through an online electrochemical mass spectrometer system is in principle possible, but must be approached with critical care to ensure proper identification of gas species and their corresponding electrochemical phenomena and time of formation.
Experimental
1) Setup
For mass detection, a HiQuad QMA410 crossbeam mass spectrometer (MS) from Pfeiffer Vacuum® with a continuous secondary electron multiplier detector was used. The MS had a m/z scan range of 1–128 amu. For electrochemical investigations, a Biologic® VSP300 potentiostat and the Software EC-Lab® (version V11.43) were used. Data assignment between the MS ion current signal and the electrochemical recording was done using Origin® (version 2019) and the local time in both text files. The electrochemical cell (see Fig. 6) is made from stainless steel. All connections are made by using stainless steel Swagelok connectors (Swagelok®, USA). The cell is connected via a stainless steel crimped capillary leak open (Vacuum Technology Incorporation®, USA) to the MS [20, 25]. The crimped capillary provides a leak rate of 10–5 mbar dm3 s−1 and has a 0.5 µm stainless steel cup filter, which provides a continuous laminar flow from ambient pressure to the UHV regime.

Scheme of the electrochemical cell and the LIC configurations
2) Materials
The negative electrode was prepared by stirring a 5.0 wt.% solution of PVDF (Solef® 5140, Solvay Belgium) together with N-methyl-2-pyrrolidone (NMP, anhydrous, 99.5% Sigma-Aldrich) for 12 h at room temperature. Then, 2% super C65 (IMERYS®, Switzerland) was added and the mixture was stirred for 30 min in a planetary orbital mixer with 2000 rpm (Thinky Are 250, USA). Subsequently, 93 wt.% graphite powder (SMG-A5, Hitachi, Japan) was added and the solution was mixed for 10 min with 2000 rpm. The slurry was coated on either a stainless steel mesh (0.212/0.090 mm, Spoerl KG, Germany) using a 150 µm gap applicator or on a copper foil (10 µm thickness Iljin, South Korea). In both cases, an automatic film applicator (TQC Sheen) was used. The coating was first dried in a ventilated oven at 100 °C before punching out in 16 mm disks. The positive electrodes were produced using 96.0 wt.% NMC811 (Umicore, Belgium), together with 0.2 wt.% BM-730H (Zeon, Japan), 1.8 wt. % Solef5140 and 2.0 wt.% LITXHP (Cabot Corporation, USA). The NMP containing slurry was coated on a aluminum foil (15 µm thickness Hydro, Norway). The dried electrodes were punched out in 14 mm disks. Positive and negative electrodes were separated using a Whatman separator (Whatmann PLC, United Kingdom), which was soaked with 200 µL electrolyte. The liquid electrolyte was a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) in 1:1 ratio (v/v) together with 1 M of LiPF6 (Sigma Aldrich, Germany).
3) Cell preparation and electrochemical procedure
For all described experiments, the cell parts and electrodes were dried within a drying cabinet (Büchi oven) at 120 °C for at least 12 h and subsequently transferred into an argon filled glovebox (MBraun®; O2 and H2O < 1 ppm). Cells were built by placing a positive electrode on the bottom of the cell, followed by the separator, and adding 200 µL electrolyte. Finally, the sandwich was covered with the negative graphite containing electrode sheet (either foil or mesh) and an additional iron mesh for contacting the electrode stack.
The cell was sealed and connected via the crimped capillary leak toward the MS. Due to the high vapor pressure and gas phase equilibrium rearrangement, the cell was rested for 4 h under open circuit conditions (mesh electrodes) or 11 h (foil electrodes). Then, chronoamperometry was carried out. For the time variation of the mesh-coated positive electrodes, the following procedure was carried out: A potential of 4.2 V was applied for 1 s, 2 s, 3 s, 4 s, and 5 s, respectively. After each potential pulse application, the electrode system was kept under OCV conditions for one hour, while MS recording was done. The procedure for foil coated electrodes was as follows: 1 s (0.5 h OCV), 2 s (0.5 h OCV), 3 s (0.5 h OCV), 4 s (0.5 h OCV), 5 s (1 h OCV), and 7.5 s (1 h OCV). For the potential variation with a four second pulse duration, the following procedure was carried out: 2.0 V (45 min OCV), 2.5 V (45 min OCV), 3.0 V (45 min OCV), 3.5 V (45 min OCV), 4.0 V (45 min OCV), and 4.5 V (45 min OCV).
4) Data processing
To avoid signal fluctuations due to minor pressure and temperature changes, the collected raw MS data were divided by the signal intensity of the 36Argon (m/z = 36) isotope and further labeled as Ix/I36Ar. Furthermore, a background correction was carried out by subtracting the value for each gas species at the end of the OCV phase. As some gasses may overlay with the signals from the gasses of interest, the following equations for the calculation were used. Hydrogen was measured using the m/z = 2 trace, whereas ethylene was measured using the m/z = 26 trace. Carbon dioxide used the m/z = 44 values and oxygen used the m/z = 32 value. Carbon monoxide had to re-calculated as ethylene and carbon dioxide showed a strong contribution on the same mass channel. The intensity of CO was recalculated using the formula: I28CO = I28 − 0.14 × I44 − (1/0.63 × I26) [19]. The formula accounts for the typical fragmentation contributions of carbon dioxide (0.14 of the base peak current) and ethylene (1/0.63 of the base peak current) to the mass channel 28. By subtracting both contributing components, the corrected signal intensity due to CO evolution can be estimated.
Data availability
All relevant data are included in the manuscript.
References
Visual Capitalist. Visualizing 25 years of lithium production by country, US, Vancouver. https://www.visualcapitalist.com/ Accessed 23 Feb 2023
Lundström R, Berg EJ (2011) J Power Sources 485:229347
Mattinen U, Klett M, Lindbergh G, Wreland R (2020) J Power Sources 477:228968
Misiewicz C, Lundström R, Ahmed I, Lacey M, Brant W, Berg EJ (2023) J Power Sources 554:232318
Sångeland C, Sun B, Brandell D, Berg EJ, Mindemark J (2011) Batter Supercaps 4:785
Teng X, Zhan C, Bai Y, Ma L, Liu Q, Wu C, Wu F, Yang Y, Lu J, Amine K (2015) ACS Appl Mater Interfaces 7:41
Rowden B, Garcia-Araez N (2020) Energy Rep 6:5
Eidenschink J, Bagherimetkazini S, Matysik FM (2022) Monatsh Chem 153:775
Iffelsberger C, Wert S, Pumera M, Matysik FM (2021) ACS Appl Mater Interfaces 13:30
Herl T, Heigl N, Matysik FM (2018) Monatsh Chem 149:1685
Cindric M, Matysik FM (2015) TrAC Trends Anal Chem 70:122
He M, Castel E, Laumann A, Nuspl G, Novák P, Berg EJ (2015) J Electrochem Soc 162:6
Kitz P, Lacey M, Novák P, Berg EJ (2020) J Power Sources 477:228567
Imhof R, Novak P (1998) J Electrochem Soc 145:1081
McCloskey B, Bethune D, Shelby R, Girishkumar G, Luntz A (2011) J Phys Chem Lett 2:10
Tsiouvaras N, Meini S, Buchberger I, Gasteiger H (2013) J Electrochem Soc 160:3
Anouti M, Dougassa YR, Tessier C, El Ouatani L, Jacquemin J (2012) J Chem Thermodyn 50:71
Raghibi M, Xiong B, Phadke S, Anouti M (2020) Electrochim Acta 362:137214
Strehle B, Solchenbach S, Metzger M, Schwenke U, Gasteiger H (2017) J Electrochem Soc 164:12
Mott Corporation Porous Metal Products, Plainville, US. https://mottcorp.com/de/ Accessed 20 Jan 2023
Solchenbach S, Metzger M, Egawa M, Beyer H, Gasteiger H (2018) J Electrochem Soc 165:13
Guégen A, Streich D, Minglong H, Mendzed M, Chesnau F, Novák P, Berg E (2016) J Electrochem Soc 163:6
Jung R, Metzger M, Maglia F, Stinner C, Gasteiger H (2017) J Phys Chem Lett 8:19
Barani weather stations and meteorological sensors (2003), Seattle US. https://www.baranidesign.com/ Accessed 24 Feb 2023
Vacuum Technology Incorporation (1985), Oak Ridge, TN, US https://vacuumtechnology.com/ Accessed 20 Jan 2023
Acknowledgements
The authors acknowledge the German Federal Ministry for Economics and Technology, the IPCEI-EuBatIn (Important Projects of Common European Interests, grant No. 16BZF205, 16BZF200) and the Bavarian state ministry for economics, regional development, and energy, for their financial funding support. Furthermore, we thank Jan-Niklas Schaper, Matthias Schroeder, Sabrina Schönemeyer and Verena Peters for their proof of reading as well as Johannes Wandt and Erik Berg for their invaluable support by setting up the OEMS system.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scharf, J., Matysik, FM. Study of the response characteristics of an online electrochemical mass spectrometry system for gas analysis of lithium-ion cells using chronoamperometry. Monatsh Chem 154, 1025–1033 (2023). https://doi.org/10.1007/s00706-023-03094-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-023-03094-z