
Abstract
The conversion of benzophenone methylhydrazone with bromine and pyridine into 1-[(methyldiazenyl)diphenylmethyl]pyridin-1-ium bromide followed by the solvolysis with methanol provides a facile synthesis of 1-(methoxydiphenylmethyl)-2-methyldiazene. The acid-induced hydrolysis of this N,O-ketal releases the methyldiazenyl moiety as the putative intermediate methyldiazenium ion. Reacting as a heterodienophile, methyldiazenium ion is intercepted with 1,3-dienes in [4 + 2+] cycloaddition reactions affording 1-methyl-1,2,3,6-tetrahydropyridazine Diels–Alder products which are transformed into stable and isolated N-benzoyl derivatives.
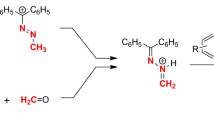
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In 1925, Diels, Blom, and Koll [1] published the result of the reexamination of the earlier reported reaction [2] of cyclopentadiene with diethyl (E)-diazene-1,2-dicarboxylate (diethyl azodicarboxylate, DEAD, EtO2C–N = N–CO2Et) and proved the product structure as diethyl 2,3-diazabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate. Apparently, this tetrahydropyridazine product—resulting from the reaction of a 1,3-diene with an azo compound as the dienophile—is the first example of a 6-membered (heterocyclic) adduct reported at the dawn of the era of [4 + 2] cycloaddition reactions that later have been coined the term ‘Diels–Alder reactions’.
Already at this early stage, a common feature of ‘normal electron demand’ Diels–Alder reactions, namely the activation of the dienophile double bond by the attachment of electron-withdrawing groups (EWG) had been fortuitously practiced. This concept applies especially to diazenes (azo compounds) as heterodienophiles [3].
Another common way of efficiently boosting the dienophilicity of a π-system is the incorporation of a positive charge at the nitrogen atom of the dienophile, e.g., converting imines (> C = N–) into iminium ions (> C = N+ <) [4]. This method of activating dienophiles has been applied also to diazenes, but only few examples of diazenium ions (–N = N+ <) reacting as dienophiles can be found in the literature. Urry et al. [5, 6] described the preparation of the stable 1,1-dimethyldiazenium bromide (Me2N+ = NH Br–) and its reaction as dienophile with 1,3-dienes to form 1,1-dimethyl-1,2,3,6-tetrahydropyridazin-1-ium bromides. This type of [4 + 2+] cycloaddition reaction has been further explored by Zelenin et al. [7, 8]. Unactivated 1,2-disubstituted diazenes (azo compounds, R–N = N–R, R = alkyl, aryl) are known to be inert toward conjugated dienes even in the presence of protic acids. A rare exception has been reported by Nelsen et al. [9, 10]. The bicyclic, thus constitutionally Z-configurated diazenes, 2,3-diazabicyclo[2.2.1]hept-2-ene and 2,3-diazabicyclo[2.2.2]oct-2-ene, upon protonation with tetrafluoroboric acid and formation of the conjugate acids, i.e. diazenium ions, readily undergo [4 + 2+] cycloaddition with 1,3-cyclohexadiene forming the corresponding tetracyclic adducts.
Monosubstituted diazenes—frequently postulated as reaction intermediates—can be prepared under stringent conditions. Physical and chemical properties of alkyl- and aryldiazenes (R–N = NH, R = alkyl, aryl) both in substance and in solutions have been studied [11,12,13]. A common feature of monosubstituted diazenes is their fast decomposition. Major decomposition products of aryldiazenes (Ar–N = N–H) in neutral and in basic media are mostly the parent hydrocarbon ArH and N2 [11]. Exposed to aqueous acids, aryldiazenes decompose instantaneously although in a different way.
Aryldiazenyl group containing N,O-ketals (hemiaminal ethers, ArN = NC(Ph2)–OR)—a class of azo compounds first described by Huisgen and Koch [14]—are highly sensitive toward acids. Treatment of aryldiazenyl hemiaminal ethers with aqueous acid affords the hydrolysis products ROH, Ph2C = O, and the putative intermediates ArN = NH. Taking into account the reaction conditions, the conjugate acids, i.e. aryldiazenium ions (ArNH+ = NH and/or ArN = NH2+) may have to be considered as well. The formation of those intermediates has been indicated by the isolation of some of their redox products [14].
Similarly, a convenient source for generating aryldiazenium ions is the facile acid-induced hydrolysis of aminals (N,N-ketals) composed of an aryldiazenyl group as one of the two nitrogen containing groups (Ar–N = N–C(R2)–N < , R = alkyl or aryl) [15, 16]. In the absence of added scavengers only traces of the parent arene of the aryldiazenyl group have been detected. Instead, products derived from the aryldiazenyl group, ArNH2, ArN2+, ArNHNH2, and ArN3 have been isolated as such or as derivatives [15,16,17,18]. These disproportionation products are congruent with the nitrogen containing products NH3·H2SO4, N2, H2NNH2·H2SO4, and HN3 obtained from the decomposition of diazene-1,2-dicarboxylic acid (HO2CN = NCO2H) with sulfuric acid resulting from the disproportionation of the parent diazene (diimide, diimine, HN = NH) [19, 20] and—considering the reaction conditions—with the conceivable participation of the diazenium ion (H2N+ = NH).
In the presence of suitable scavangers like 1,3-dienes, aryldiazenium ion intermediates (ArNH+ = NH and ArN = NH2+) can be intercepted as dienophiles undergoing [4 + 2+] cycloaddition reactions furnishing tetrahydropyridazines [15, 16, 21,22,23,24]. More recently, the synthesis of pyridazinium salts has been reported resulting from the reaction of furan with aryldiazenium ion intermediates generated by the addition of acid to solutions of aryldiazenes [25, 26].
The decomposition of the smallest alkyldiazene, methyldiazene (CH3N = NH) with aqueous acids HA (A = OSO3H, Cl) has been thoroughly examined by Ackermann et al. [12]. Beside small amounts of CH4 a number of isolated products arise from nucleophilic displacement reactions at the carbon atom of the 1-methyldiazen-1-ium ion intermediate (CH3NH+ = NH). Diazene (HN = NH) as the leaving group undergoes disproportionation to N2 and H2NNH2, the latter, in turn, giving rise to methylated products CH3NHNH2 and (CH3)2NNH2 in addition to CH3OH and CH3A derived from the aqueous acids employed.
Aryldiazenyl group containing N,O-ketals—prepared by various methods [14, 27,28,29,30]—have been successfully utilized to generate aryldiazenium ion dienophiles to react with 1,3-dienes yielding Diels–Alder products [21,22,23,24]. An analogous approach was designed for an exploratory investigation of methyldiazenium ion and the interception of this electron-deficient dienophile by 1,3-dienes as scavengers in [4 + 2+] cycloaddition reactions.
Results and discussion
1-(Alkoxydiphenylmethyl)-2-methyldiazenes 3 (Scheme 1) were envisaged suitable precursors for producing methyldiazene (4) and the conjugate acid, methyldiazenium ion (4H+) in the course of an acid-induced hydrolysis.

Preparation of methyldiazenyl hemiaminal ethers 3
The preparation of the N,O-ketals 3a, 3b was accomplished in analogy to the synthesis of 1-(alkoxydiphenylmethyl)-2-aryldiazenes [29, 30].
For the preparation of the methyldiazenyl hemiaminal methyl ether 3a (Scheme 1) a solution of benzophenone methylhydrazone 1 in dichloromethane was added to a solution of bromine in pyridine. Subsequent addition of diethyl ether precipitated a mixture of salts, 1-[(methyldiazenyl)diphenylmethyl]pyridin-1-ium bromide (2) accompanied by pyridinium bromide. The salt mixture was filtered off and had to be used for the next step without delay because pyridinium salt 2 decomposed readily, in particular, in the presence of moist solvents, presumably by undergoing a facile 1,4-elimination of pyridinium bromide. Solvent-free samples of the inseparable pyridinium salts are stable for several days. A sample of the salt mixture was dissolved in acetone-d6 and permitted to obtain the 1H NMR spectrum displaying a singlet at δ = 4.13 ppm indicative of the methyldiazenyl group of 2. The salt mixture was dissolved in anhydrous methanol, anhydrous diethyl ether was added and precipitated pyridinium bromide. It is important to quickly remove pyridinium bromide by filtration to prevent its reaction as a protic acid with the hemiaminal ether 3a. From the filtrate (E)-1-[methoxy(diphenyl)methyl]-2-methyl-diazene (3a) was isolated in good yield. Analogously, treatment of the pyridinium salt mixture of 2 with anhydrous ethanol provided (E)-1-[ethoxy(diphenyl)methyl]-2-methyldiazene (3b). Methyldiazenyl hemiaminal ethers 3 (like aryldiazenyl hemiaminal ethers [14, 27,28,29,30]) are acid sensitive but pure and solvent-free samples can be stored for long periods.
Reaction of methyldiazenyl hemiaminal methyl ether 3a with 1,3-dienes 5 in the presence of aqueous tetrafluoroboric acid
The acid-induced hydrolysis of 1-(methoxydiphenylmethyl)-2-methyldiazene (3a) was envisaged to form—in situ—methyldiazene (4) and the conjugate acid 4H+ (Scheme 2). As depicted, the structure of 1-methyl-diazen-1-ium ion (4H+) is in keeping with the previously used formulation [12]. Intermediate 4H+ was expected to react as an electron-deficient dienophile and when generated in the presence of 1,3-dienes would participate in [4 + 2+] cycloaddition reactions.

Under ice-cooling, a solution of the hemiaminal ether 3a in acetonitrile was added to a solution of aqueous tetrafluoroboric acid and an excess of 2,3-dimethyl-1,3-butadiene (5a) in acetonitrile and. The reaction conditions intended to ensure that diene 5a is available in excess for the reaction with the methyldiazenium ion (4H+). Moreover, the purpose of the slow addition of the hemiaminal ether 3a to the aqueous acid solution was to maintain a low concentration of methyldiazene (4) and the conjugate acid 4H+, thus minimizing the risk of decomposition that these intermediates are inclined to suffer [12].
Work-up of the reaction included removal of excessive diene and the solvents, treatment of the residue with sodium hydroxide, repeated extraction of the residue with diethyl ether, removal of the solvent, addition of hydrochloric acid to the residue, and another extraction with ether. The two layers were separated, the aqueous phase was basified with sodium hydroxide but the isolation of the [4 + 2+] cycloadduct, 1,4,5-trimethyl-1,2,3,6-tetrahydropyridazine (6a) was not attempted because of the anticipated air sensitivity of the product. Instead, a Schotten-Baumann procedure [31, 32] with benzoyl chloride produced the stable N-benzoyl derivative. Purification by column chromatography and recrystallisation afforded pure crystals of 1-benzoyl-2,4,5-trimethyl-1,2,3,6-tetrahydropyridazine (7a) in 36% yield.
The ether phase—after separation from the aqueous acidic layer—was checked by thin layer chromatography (tlc) exhibiting two major spots assigned to benzophenone and a by-product that was identified by comparison with authentic N-(diphenylmethylene)-3,4-dimethyl-5,6-dihydropyridin-1(2H)-amine. The preparation of such alternative Diels–Alder cycloadducts as the sole products from N,O-ketal 3a and 1,3-dienes 5 albeit under different reaction conditions will be presented and discussed in a forthcoming communication [33].
The reaction of 2-methyldiazenyl hemiaminal ether 3a with (E)-penta-1,3-diene (trans-piperylene, 5b) gave rise to two [4 + 2+] cycloadducts, 1,3-dimethyl-1,2,3,6-tetrahydropyridazine (6b) and 1,6-dimethyl-1,2,3,6-tetrahydropyridazine (6c) which were converted into the N-benzoyl derivatives. The two regioisomers, 1-benzoyl-2,6-dimethyl-1,2,3,6-tetrahydropyridazine (7b) and 1-benzoyl-2,3-dimethyl-1,2,3,6-tetrahydropyridazine (7c) were separated by column chromatography and obtained as pure crystals in low yield, (7.8% and 5.2%, respectively). The structures of 7b and 7c have been confirmed by their 1H and 13C NMR spectra including DEPT and COSY (Supplementary Materials). The yield ratio of 3:2 in favor of the ortho-regioisomer 7c reflects the similar isomer ratio of regioisomers obtained from the [4 + 2+] cycloaddition of aryldiazenium ions with penta-1,3-diene 5b [24].
By the same procedure, the reaction of N,O-ketal 3a was carried out with 1,3-cyclohexadiene (5d) yielding the [4 + 2+] cycloadduct 2-methyl-2,3-diazabicyclo[2.2.2]oct-5-ene (6d), which was isolated as the N-benzoyl derivative, 2-benzoyl-3-methyl-2,3-diazabicyclo[2.2.2]oct-5-ene (7d) in 23% yield.
Conceivable pathways for the formation of 1-methyldiazen-1-ium ion (4H.+)
The reaction of methyldiazenyl N,O-ketal 3a with aqueous acid generating the putative dienophile 1-methyldiazen-1-ium ion (4H+) can be plausibly discussed via two conceivable pathways (Scheme 3).

Protonation of the hemiaminal ether 3a at the methoxy group (O-protonation, route a, Scheme 3) entails the dissociation of methanol and formation of the 1-(diphenylmethylene)-2-methyldiazen-1-ium ion (8+), which upon reaction with water gives rise to the conjugate acid 9H+ of (methyldiazenyl)diphenylmethanol. Dissociation of the O-protonated hemiaminal 9H+ affords benzophenone and the 1-methyldiazen-1-ium ion (4H+).
Alternatively, protonation of the methyldiazenyl moiety of 3a (N-protonation, route b, Scheme 3) gives rise to two conjugate acids 3aH+ depending on the protonation site. Considering the general lack of dienophilicity of 1,2-dialkyl-substituted diazenium ions (vide supra for a notable exception [9, 10]), intermediates 3aH+ can be disregarded as dienophiles. Instead, subsequent dissociation of either N-protonated 2-methyldiazenyl hemiaminal ether 3aH+ appears to be more likely, forming methyldiazene (4) (formulated as such and as the 2-methyldiazen-2-ium-1-ide isomer) together with (diphenylmethylene)(methyl)oxonium ion (10+). The latter intermediate 10+ reacting with water produces methanol and benzophenone while methyldiazene 4 (either form) is converted into 1-methyldiazen-1-ium ion (4H+).
Spectra
The structures of the heterocyclic products 7 have been identified based on their spectral data (1H and 13C NMR spectra in Supplementary Materials). The 1H and 13C NMR spectra of 7d are complex and exhibit broadened signals at ambient probe temperature reminiscent of those of similar bridged and N,N-disubstituted 1,2,3,6-tetrahydropyridazines [34, 35]. The temperature dependence of the NMR spectra of symmetrically 2,3-disubstituted 2,3-diazabicyclo[2.2.2]-oct-5-enes has been attributed to various effects [34, 35]: hindered nitrogen inversion, ring twisting, and ring inversion for 2,3-dimethyl-substituted tetrahydropyridazines as well as restricted rotation about the N–CO2Me bond for 2,3-dicarbomethoxy-substitited bridged tetrahydropyridazines [34, 35]. Thus, the complex and temperature dependent NMR spectra of 7d (two different groups—benzoyl and methyl—attached to the nitrogen atoms of the tetrahydropyridazine ring) are not unexpected. A detailed analysis of the NMR spectra of 7d was beyond the scope of this synthesis-oriented work.
Conclusion
This communication presents the preliminary and exploratory work on the acid-induced hydrolysis of methyldiazenyl hemiaminoketal methyl ether (3a), providing a convenient source for generating methyldiazenium ion (4H+) in situ. As anticipated, intermediate 4H+ acting as an electron-deficient heterodienophile was successfully intercepted with 1,3-dienes 5 in [4 + 2+] cycloaddition reactions affording the Diels–Alder products 1,2,3,6-tetrahydropyridazines 6 which were converted into the stable N-benzoyl derivatives 7. The low yields of the isolated N-benzoyl derivatives 7 may be due to the known decomposition reactions of the intermediates methyldiazene (4) and methyldiazenium ion (4H+) [12] in competition with the interception by dienes 5 forming the Diels–Alder cycloadducts 6.
Experimental
Most reagents are commercially available from Fluka or Merck. The 1,3-dienes had a purity of > 97%; 2,3-dimethyl-1,3-butadiene and 1,3-cyclohexadiene were stabilized with 0.1% hydroquinone and 0.1% 2,6-di-tert-butyl-4-methylphenol, respectively. All solvents were freshly distilled. Petroleum ether refers to the fraction of the boiling range 40–60 °C. Solvents were evaporated using a rotary evaporator Vapsilator (Chemophor) at ca 2 kPa.
Thin layer chromatography (tlc) analyses were carried out on pre-coated polyester sheets with a silica gel layer, 4 × 8 cm (POLYGRAM SIL G UV254, Macherey–Nagel). Column chromatographic separations were performed as low-pressure liquid chromatography (LPLC): glass columns (length 45 cm, i.d. 2.6 or 3.7 cm), silica gel 40–60 mm, 250 g (MERCK Kieselgel 60), external pressure 150–250 kPa; fractions of 10 cm3 were collected.
Melting points were determined on a Kofler hot stage microscope (Thermovar-Reichert). The spectroscopic data have been obtained with the following instruments: Beckman AccuLab 4 (IR), Gilford Spectrophotometer 250 (UV–Vis), JEOL JNM- PMX 60 (lH NMR, 60 MHz) and Bruker AM 300 (1H and 13C NMR, 300.13 and 75 MHz, respectively), Varian MAT 44S (MS).
Elementary microanalyses (C, H, N) were performed by Dr. J. Zak at the Institute of Physical Chemistry of the University of Vienna. The results are in good agreement with the calculated values.
1-(Diphenylmethylene)-2-methylhydrazine (benzophenone methylhydrazone, 1)
To a solution of methylhydrazine (10 cm3, 190 mmol) and benzophenone (27.30 g, 150 mmol) in cyclohexane (200 cm3) was added acetic acid (2.0 cm3). After refluxing the mixture for 80 h the solvent and excessive methylhydrazine were distilled off. The residue was distilled in vacuo (4 Pa, boiling range 115–140° C) furnishing yellowish crystals 1 (28.8 g, 95%). M.p.: 42–43 °C (lit. 42–43 °C [36]); 1H NMR (60 MHz, CDCl3): δ = 3.05 (s, 3H, NCH3), 5.00 (br, 1H, NH), 7.28–7.76 (m, 10H, 2 C6H5) ppm.
1-[Methoxy(diphenyl)methyl]-2-methyldiazene (3a, C15H16N2O)
To ice-cold pyridine (25 cm3, 310 mmol) was added with stirring a solution of bromine (5.60 g, 35 mmol) in dichloromethane (30 cm3). Thereafter, a solution of benzophenone methylhydrazone (1, 7.36 g, 35 mmol) in dichloromethane (20 cm3) was added dropwise within 6 min, and the reaction mixture was stirred at room temperature for 30 min. Addition of anhydrous ethyl ether (150 cm3) precipitated a mixture of 1-[(methyldiazenyl)diphenylmethyl]pyridin-1-ium bromide (2) and pyridinium bromide. The salt mixture was filtered off and washed with diethyl ether. A sample of the pyridinium salts mixture was dissolved in CD3CN and showed a transient signal at 4.13 ppm indicative of the NCH3 group in 2. The inseparable, slightly pink mixture of the pyridinium salts was dissolved in ice-cold methanol (40 cm3, 989 mmol). To the resulting solution was added with stirring anhydrous diethyl ether (100 cm3), and after 1 h more diethyl ether (100 cm3) was added. Stirring was continued for 1 h, and the colorless, crystalline precipitate of pyridinium bromide was filtered off. The filtrate was washed with water and dried (MgSO4). The solvents were removed in vacuo leaving behind pure (by tlc), slightly yellow crystals 3a (6.80 g, 79%). Compound 3a is acid sensitive but pure, solvent-free samples can be stored. M.p.: 56–57 °C (methanol); UV (CH3CN): λmax (log ε) = 251 (4.22), 330 (2.01) nm; 1H NMR (60 MHz, CDCl3): δ = 3.28 (s, 3H, OCH3), 4.00 (s, 3H, NCH3), 7.23–7.80 (m, 10H, 2 C6H5) ppm.
1-[Ethoxy(diphenyl)methyl]-2-methyldiazene (3b, C16H18N2O)
The same procedure as described above was applied, except for employing ethanol (150 cm3, 2.57 mol) and a reaction time of 3 h to provide pure (by tlc), slightly yellow crystals 3b (3.38 g, 38%). M.p.: 42–43 °C (ethanol); UV (CH3CN): λmax (log ε) = 251 (4.24), 327 (2.05) nm; 1H NMR (60 MHz, CDCl3): δ = 1.18 (t, J = 7 Hz, 3H, CCH3), 3.30 (q, J = 7 Hz, 2H, OCH2), 3.80 (s, 3H, NCH3), 7.07–7.70 (m, 10H, 2 C6H5) ppm.
1-Benzoyl-2,4,5-trimethyl-1,2,3,6-tetrahydropyridazine (7a, C14H18N2O)
To an ice-cooled and stirred mixture of 2,3-dimethylbutadiene (5a, 20 cm3, 177 mmol), acetonitrile (30 cm3), and hydrogen tetrafluoroborate (2.5 cm3, 50% in water) was added dropwise a solution of 1-[methoxy(diphenyl)methyl]-2-methyldiazene (3a, 3.60 g, 15 mmol) in acetonitrile (20 cm3) within 12 min. Upon continued stirring at room temperature for 50 min the reaction mixture turned into a clear slightly yellow solution. The solvent and excess of diene 5a were removed in vacuo. The residual brown oil (8.02 g) was treated with 2 N sodium hydroxide (30 cm3) and repeatedly extracted with diethyl ether (6 × 50 cm3). The combined ether extract was dried (MgSO4) and the solvent removed in vacuo. The residual yellow oil (4.14 g) upon addition of 2 N hydrochloric acid (40 cm3) was repeatedly extracted with diethyl ether. A tlc check (diethyl ether/petroleum ether 1:2) of the ether extract showed two major spots: benzophenone (Rf = 0.75) and a spot (Rf = 0.83) matching the Rf of authentic N-(diphenylmethylene)-3,4-dimethyl-5,6-dihydropyridin-1(2H)-amine [33]. The aqueous phase was basified with 2 N sodium hydroxide (60 cm3), and benzoyl chloride (3.0 cm3, 26 mmol) was added. The reaction mixture was stirred for 12 h and then extracted with diethyl ether (250 cm3). The ether phase was washed with water until neutral and dried (MgSO4). Removal of the solvent in vacuo left an oily, semicrystalline residue (1.74 g), which was subjected to LPLC (silica gel 250 g, diethyl ether/petroleum ether 1:2). Fractions 57–90 contained the colorless, crystalline product 7a (1.18 g, 34% after two recrystallizations from pentane). M.p.: 58–59 °C; Rf = 0.25 (diethyl ether/petroleum ether 1:2); IR (KBr): \(\overline{v}\) = 1630 cm–1; 1H NMR (300.13 MHz, CDCl3): δ = 1.62 (s, 3H, 4-CH3), 1.71 (s, 3H, 5-CH3), 2.50 (s, 3H, 2-NCH3), 2.71 (br d, 1H, 3-HA), 3.63 (br, 2H, 3-HB, 6-HA), 4.42 (br d, 1H, 6-HB), 7.25–7.40 (m, 3H, C6H5), 7.50–7.60 (m, 2H, C6H5) ppm; 13C NMR (75.47 MHz, CDCl3): δ = 15.8, 15.9 (4,5-CH3), 38.8 (6-CH2), 41.7 (2-NCH3), 57.3 (3-CH2), 120.8, 121.1 (4,5-CH), 127.5, 129.5, 136.3 (C6H5), 171.0 (C = O) ppm; EI-MS: m/z (%) = 230 (2.75, M+), 125 (22.82), 105 (9.00), 77 (12.12), 55 (15.07), 43 (100), 42 (67.78).
1-Benzoyl-2,6-dimethyl-1,2,3,6-tetrahydropyridazine (7b, C13H16N2O) and 1-benzoyl-2,3-dimethyl-1,2,3,6-tetrahydropyridazine (7c, C13H16N2O)
Under the condition of the procedure described above a mixture of (3E)-penta-1,3-diene (trans-piperylene, 5b) (15.0 cm3, 150 mmol), acetonitrile (30 cm3), hydrogen tetrafluoroborate (2.0 cm3, 50% in water) was combined with a solution of 3a (4.3 g, 18 mmol) in acetonitrile (25 cm3). The reaction mixture was stirred for 15 min at ambient temperature. After removal of the solvent and excess diene 5b in vacuo, 2 N sodium hydroxide (30 cm3) was added to the residual orange-brown oil (5.40 g). The aqueous layer was repeatedly extracted with ether (6 × 50 cm3). The extract was dried (MgSO4), the solvent was distilled off in vacuo furnishing a yellow-orange oil with colorless needles (4.03 g) that was treated with 2 N hydrochloric acid (40 cm3) and any undissolved material was removed by extraction with ether. Upon basification of the acidic aqueous layer with 2 N sodium hydroxide and addition of benzoyl chloride (4.0 cm3, 34 mmol) the mixture was stirred at ambient temperature for 12 h. After extraction with diethyl ether, drying of the extract (MgSO4), and removal of the solvent a brown oil (2.33 g) remained. Separation of the reaction mixture by LPLC (silica gel (250 g), diethyl ether/petroleum ether 1:1) afforded three major fractions: (a) Fractions 1–15 contained benzoic anhydride (1.25 g). M.p.: 39–40 °C (lit. 43 °C [37]); Rf = 0.65 (diethyl ether/petroleum ether 1:1); IR (KBr): \(\overline{v}\) = 1760, 1700 cm–1 (C = O); EI-MS: m/z (%) = 226 (5.94, M+), 105 (100). (b) Fractions 35–50 afforded yellowish crystals 7b (0.20 g, 5%). M.p.: 87–89 °C (pentane); Rf = 0.45 (diethyl ether/petroleum ether 1:1); IR (KBr): \(\overline{v}\) = 1610 cm–1 (C = O); 1H NMR (300.13 MHz, CDCl3): δ = 1.49 (d, J = 7 Hz, 3H, 3-CH3), 2.76 (s, 3H, 1-CH3), 3.01 (br d, 1H, 6-CHA), 3.62 (br d, 1H, 6-CHB), 4.81 (br s, 1H, 3-CH), 5.70–5.90 (m, 2H, CH = CH), 7.30–7.40 (m, 3H, C6H5), 7.50–7.55 (m, 2H, C6H5) ppm; 13C NMR (75.47 MHz, CDCl3): δ = 20.7 (3-CH3), 45.0 (1-CH3), 45.3 (3-CH), 51.7 (6-CH2), 120.6, 127.0, 127.5, 128.3, 129.1, 137.1 (C6H5), 171.1 (C = O) ppm; EI-MS: m/z (%) = 216 (4.39, M+), 111 (63.71), 110 (21.48), 105 (18.75), 95 (7.60), 77 (21.92), 55 (11.18), 43 (100), 42 (48.20), 41 (10.07). (c) Fractions 56–76 provided colorless crystals 7c (0.30 g, 8%). M.p.: 76–77 °C (pentane); Rf = 0.33 (diethyl ether/petroleum ether 1:1); IR (KBr): \(\overline{v}\) = 1605 cm–1 (C = O); 1H NMR (300.13 MHz, CDCl3): δ = 1.02 (d, J = 7 Hz, 3H, 3-CH3), 2.63 (s, 3H, 2-CH3), 3.08 (br s, 1H, 3-CH), 3.73 (br d, 1H, 6-CHA), 4.75 (br d, 1H, 6-CHB), 5.70–5.90 (m, 2H, CH = CH), 7.30–7.40 (m, 3H, C6H5), 7.49–7.55 (m, 2H, C6H5) ppm; 13C NMR (75.47 MHz, CDCl3): δ = 20.1 (3-CH3), 35.1 (6-CH2), 42.2 (1-CH3), 57.6 (3-CH), 121.1, 127.1, 127.4, 127.7, 129.1, 136.6 (C6H5), 172.1 (C = O) ppm; EI-MS: m/z (%) = 216 (2.36, M+), 185 (4.37), 111 (76.10), 110 (25.90), 105 (28.59), 95 (21.66), 77 (26.84), 76 (10.23), 55 (14.76), 43 (100), 42 (46.63), 41 (8.61)..
2-Benzoyl-3-methyl-2,3-diazabicyclo[2.2.2]oct-5-ene (7d, C14H16N2O)
Under the condition of the preceding procedure a solution of 3a (2.0 g, 8.3 mmol) in acetonitrile (15 cm3) was slowly added with stirring to an ice-cooled mixture of 1,3-cyclohexadiene (5d, 9.0 cm3, 94 mmol), acetonitrile (15 cm3), and hydrogen tetrafluoroborate (2.0 cm3, 50% in water). Stirring was continued until the reaction mixture has warmed up to room temperature and became a clear, yellowish solution. The solvents and excess diene 5d were stripped off and to the semicrystalline residue was added 2 N sodium hydroxide (30 cm3). The resulting yellow-orange solution was repeatedly extracted with diethyl ether. The combined extract was dried (MgSO4), the solvent was removed in vacuo leaving behind a yellow oil (2.15 g). The oil was dissolved in 2 N hydrochloric acid (20 cm3) and repeatedly extracted with diethyl ether. The aqueous phase was basified with 2 N sodium hydroxide (20 cm3), benzoyl chloride (3.5 cm3, 30 mmol) was added, and the mixture was stirred for 12 h. Subsequent extraction with diethyl ether (250 cm3), drying of the extract (MgSO4), and removal of the solvent afforded a slowly crystallizing brown oil (1.65 g). Purification by LPLC (silica gel (250 g), diethyl ether/petroleum ether 3:2, and collection of fractions exhibiting a tlc spot at Rf = 0.18) afforded colorless crystals 7d (0.44 g, 23%). M.p.: 105–106 °C (pentane); Rf = 0.18 (diethyl ether/petroleum ether 3:2); IR (KBr): \(\overline{v}\) = 1630 cm–1; 1H NMR (300.13 MHz, DMSO-d6, T = 342 K): δ = 1.15–1.28, 1.42–1.56, 1.85–1.95, 2.00–2.10 (mm, 4 × 1H, CH2CH2), 2.15–2.30 (br s, 3H, NCH3), 3.73–3.80 (m, 1H, NCH), 4.75–4.95 (br s, 1H, NCH), 6.43–6.48 (m, 1H, = CH), 6.62–6.67 (m, 1H, = CH), 7.40–7.60 (m, 5H, C6H5) ppm; 13C NMR (75.47 MHz, DMSO-d6, T = 303 K): δ = 21.3 (8-CH2), 23.9 (7-CH2), 43.7 (NCH3), 45.7 (4-CH), 55.5 (1-CH), 127.0, 127.6, 129.0, 130.5, 132.7, 137.7 (C6H5), 167.25 (C = O) ppm; 13C NMR (75.47 MHz, CDCl3, T = 303 K): δ = 21.9 (CH2), 24.5 (CH2), 41.8, 43.9 (NCH3), 46.2, 50.6 (4-CH), 56.3 (1-CH), 127.0, 127.6, 128.3, 129.3, 130.0, 132.2, 133.2, 137.0, 137.5 (C6H5), 166.5, 167.5 (C = O) ppm; EI-MS: m/z (%) = 228 (11.17, M+), 124 (12.12), 123 (100), 122 (40.72), 105 (31.04), 104 (10.60), 95 (77.74), 94 (21.01), 77 (36.70), 76 (13.18), 51 (15.25), 43 (85.91), 42 (38.17).
References
Diels O, Blom JH, Koll W (1925) Justus Liebigs Ann Chem 443:242
Albrecht W (1906) Justus Liebigs Ann Chem 348:31
Tietze LF, Kettschau G (1997) Hetero Diels-Alder reactions in organic chemistry. Topics in current chemistry, vol 189. Springer, Berlin
Heintzelman GR, Meigh IR, Mahajan YR, Weinreb SM (2005) Diels-Alder reactions of imino dienophiles organic reactions, vol 65. Wiley, New York, p 141
Urry WH, Kruse HW, McBride WR (1957) J Am Chem Soc 79:6568
Urry WH, Scesci P, Ikoku C, Morre DW (1964) J Am Chem Soc 86:2224
Zelenin KN, Bezhan IP (1970) Zh Org Khim 6:2206
Zelenin KN, Bezhan IP (1970) Khim Geterotsikl Soedin 1:93
Nelsen SF, Blackstock SC, Frigo TB (1984) J Am Chem Soc 106:3366
Nelsen SF, Blackstock SC, Frigo TB (1986) Tetrahedron 42:1769
Kosower EM (1971) Acc Chem Res 4:193 and literature cited therein
Ackermann MN, Hallmark MR, Hammond SK, Roe N (1972) Inorg Chem 11:3076
Craig NC, Kliewer MA, Shih NC (1979) J Am Chem Soc 101:2480
Huisgen R, Koch H-J (1955) Liebigs Ann Chem 591:200
Schantl J (1970) Monatsh Chem 101:1339
Schantl J (1977) Z Naturforsch 32b:72
Severin T, Schmitz R, Loske J, Hufnagel J (1969) Chem Ber 102:4152
Schantl JG (1996) Molecules 1:212
Diels O, Paquin M (1913) Ber Dtsch Chem Ges 46:2000
Angeli A (1910) Rend Sedute Accad Naz Lincei 19:29
Gstach H, Schantl JG (1983) Österr Chem Z 84:23
Gstach H, Schantl JG (1984) In: Abstracts of 5th International Conference on Organic Synthesis. Freiburg, Germany, p 103
Gstach H, Schantl JG (1987) Abstract of Euchem Conference on Diels-Alder Reaction. Assisi, Italy, p 15
Ho TCH (1996) Novel syntheses of pyridazine derivatives using diazenium ions and diazonium ions. Doctoral Thesis, University of Innsbruck
Fehler SK, Pratsch G, Heinrich MR (2014) Angew Chem Int Ed 53:11361
Gradl S, Köckenberger J, Oppl J, Schiller M, R. Heinrich MR (2021) J Org Chem 86:6228
Schantl J (1970) Tetrahedron Lett 11:5785
Schantl JG, Gstach H (1985) Monatsh Chem 116:1329
Gstach H, Schantl JG (1986) Synth Commun 16:741
Schantl JG, Gstach H (1987) Monatsh Chem 118:851
Schotten C (1884) Ber dtsch Chem Ges 17:2544
Baumann E (1886) Ber dtsch Chem Ges 19:3218
Gamper M, Gstach H, Schantl JG (2023) Monatsh Chem. https://doi.org/10.1007/s00706-023-03054-7
Anderson JE, Lehn JM (1967) J Am Chem Soc 89:8135
Anderson JE, Lehn JM (1968) Tetrahedron 24:123
Coleman GH, Gilman H, Adams CE, Pratt PE (1938) J Org Chem 3:99
Clarke HT, Rahrs EJ (1923) Org Synth 3:21
Funding
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
In memoriam of the dear friend and esteemed colleague Fritz Sauter.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gamper, M., Gstach, H. & Schantl, J.G. Acid-induced hydrolysis of 1-(methoxydiphenylmethyl)-2-methyldiazene: interception of methyldiazenium ion with 1,3-dienes affording 1-methyl-1,2,3,6-tetrahydropyridazine Diels–Alder products. Monatsh Chem 154, 1411–1418 (2023). https://doi.org/10.1007/s00706-023-03050-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-023-03050-x