
Abstract
Azolation in the 8-position in the purine scaffold of cAMP (adenosine 3′,5′-cyclic monophosphate) and cAMPS (adenosine 3′,5′-cyclic monophosphorothioate) provided derivatives with an azole ring directly attached to the purine via an annular azole nitrogen. Electrophilic bromination in the 8-position was followed by nucleophilic substitution with metalated azoles to afford 8-imidazo and 8-triazolo derivatives. The substrates were appropriately protected (Sp)-3′,5′-cyclic N-benzylphosphoramidate. A subsequent carbon disulfide promoted thiation reaction afforded corresponding (Rp)-8-azolo-3′.5′-cAMPS products. The reactions were stereoselective. The products as tri-n-butylammonium salts were soluble in organic solvents and were purified by chromatography. The ammonium salts were converted to sodium salts.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The heterocyclic purine framework is widely incorporated into essential biomolecular systems and constitutes an important part of medicinal chemistry. Adenine and guanine are common purine nucleobases in such frameworks as exemplified by adenosine in Fig. 1. In this report the emphasis is on development of methodology for introduction or exchange of substituents in the purine 8-position [1]. The 8-position in the purine scaffold is active in both electrophilic and nucleophilic substitution reactions [2]. 8-Bromides or 8-chlorides are useful substrates for nucleophilic substitutions affording 8-aza, 8-oxa, or 8-thia derivatives [3,4,5]. More recently, 8-nitro analogues have become available by nucleophilic displacements [6]. 8-Carbylation is achieved by Pd-promoted organometallic cross-coupling reactions with metalated carbocyclic and heterocyclic arenes [7]. Even heteroatom substituents can be introduced by cross-coupling procedures [8]. In this manner a number of 8-substituted purine skeletons have become available.

Cyclic nucleotide targets
In (RP)-adenosine-3′,5′-cyclic phosphorothioic acid (cAMPS), one of the oxygen atoms pendant from the phosphorus atom in (RP)-adenosine-3′,5′-cyclic phosphoric acid (cAMP) has been replaced by a sulfur atom in a stereoselective manner. The resulting (Rp)- and (Sp)-isomers of cAMPS are stereochemically stable and useful for various bioscreening programs associated with cyclic adenosine and cyclic guanosine monophosphates (cGMP). Both cAMP and cGMP are important secondary messenger in regulating a wide range of cell functions in response to specific hormones [9, 10]. We have reported a method for stereocontrolled preparation of 8-substituted (RP)-adenosine-3′,5′-cyclic phosphorothioic acids that possess protein kinase antagonistic activity and has a stimulating effect on the immune system [2]. In this report, we describe sp2-hybridized amino-nitrogen hinged to the 8-position in the purine skeleton by substitution reactions from the corresponding bromides or chlorides. Iodides will react in a similar manner but are less readily available intermediates. The products were cAMPS derivatives with sp2-hybridized azole nitrogen in the form of imidazole and triazoles attached to the 8-position in the nucleotide. The nucleophiles were metalated azoles. The triazolo heterocycles are π-electron deficient, and both the 1,2,3-triazoles and 1,2,4-triazoles possess low basicity. In contrast imidazole behaves as a base and nucleophile. Acyclic 8-azido derivatives were included as a less polarized species.
Results and discussion
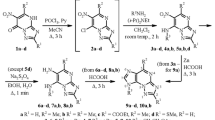
The generally low solubility of nucleosides and nucleotides in organic solvents may be partly overcome by initial conversions to amidates [2]. A solution of the (Sp)-8-bromo amidate 1 and the sodium salts of the azoles in DMF afforded the 8-azolo products. We have previously described methodology for stereoselective preparation of amidates [2]. Substitution of intermediate amidates with imidazole as sodium salt in DMF proceeded readily at elevated temperature to afford the imidazo derivative 2 (Scheme 1). 1,2,4-Triazole was reacted similarly, but the heating time was increased to 20 h because of lower reaction rate. Exclusive formation of the N-1 product 3 was observed. The proton NMR spectra were used for assignment of structures to the regioisomers. Substitution at the annular 4-nitrogen atom will yield a symmetrical triazole derivative with the same chemical shifts for the two annular triazolo protons. Different chemical shifts for the two hydrogen atoms in the azole ring in the product were observed. The unsymmetrical structure 3 was assigned to the product.

Two regioisomers were formed with 1,2,3-triazole as a reactant, viz. the symmetrical structure 4 and its isomer 5 in the ratio 3:2 (Scheme 2). The isomers were separated by flash chromatography. The symmetrical structure has the same chemical shift for both triazolo protons in the 1H NMR spectra whereas the second isomer showed two different chemical shifts for the annular triazolo protons. 1,2,3-Triazoles carrying a methyl (6) or a chloro substituent (7) in the 4-position (Scheme 2) were reacted similarly with sodium hydride as base in DMF. The 4-methyltriazole afforded the regioisomers 8 and 10 in the ratio 6:1. The 4-chlorotriazole afforded a 3:1 ratio of the regioisomers 9 and 11 that were separated by flash chromatography. The 1H shift in the methyl triazole isomers differed significantly, and isomer structures could be assigned. The major isomer was fluorescent in UV light. The chemical shifts for the triazolo proton in the two chloro isomers were almost the same. The major isomer was fluorescent and was tentatively assigned structure 8. The 5-methyl and 5-chloro triazole reactants 6 and 7 were available by literature procedures from 1-SEM-protected 1,2,3-triazol [11]. Finally, the acyclic 8-azido derivative was prepared by heating the bromide 1 with sodium azide in DMF to afford the 8-azido derivative 12. The latter is also a potential intermediate for 8-amino derived products.

Scheme 3 illustrates adaption of the Stec thiylation reaction for generation of thiophosphoric acids [12]. Initial proton abstraction from the benzylic amino group in the phosphoramidates 2–5, 8, 9 by LDA in THF at − 70 to − 40 °C afforded a lithium ylide that reacted with carbon disulfide. A sulfur atom in the intermediate becomes a nucleophile and a cyclization reaction occurs. Cleavage of the P–N bond occurs with retention of the true configuration at the phosphorus atom. The hydrophobic nature of the bulky TBDMS-protecting group attached to the 2′-hydroxy group leads to precipitation of the thioic acid from the aqueous mixture. The product was desilylated by ammonium fluoride in methanol at 45 °C to afford the target compounds (Scheme 3). The 8-azido derivative 12 was thiated in the same manner.

The deprotected products were isolated as tributylammonium salts after addition of tributylamine to the acid. The tributylammonium salts 15, 16, 20–22, 23, 24 of the products were soluble in polar organic solvents and could be purified by recrystallization or by flash chromatography on silica gel using CH2Cl2:MeOH:NBu3. The azido amidate 12 reacted in the same manner to furnish the phosphorothioic acid 24. The tributylammonium salts were converted into sodium salts 25–31 by dissolution of the ammonium salts in methanolic sodium hydroxide. Precipitation of the salts was by addition of diethyl ether (Scheme 4).

The products were subjected to T cell proliferation assays by established methodologies [2, 9, 13]. Comparison with previously reported systems indicate low specific effect by the nature of the annular heteroatom(s) in the five-membered hetarene in the purine 8-position.
Conclusion
Methods for preparation of novel 8-imidazolo and 8-triazolo derivatives of cAMP and cAMPS have been developed. Azolation in the 8-position in the purine scaffold of cAMP and cAMPS provides derivatives with annular sp2-hybridized azole amino-nitrogen attached directly to the purine ring. In the process, an appropriately silyl protected (Sp)-8-bromoadenosine 3′,5′-cyclic N-benzylphosphoramidate is aminated with a metalated azole as nucleophile, followed by stereoselective thiation at the phosphorus atom to deliver adenosine 3′,5′-cyclic phosphorothioates with retention of the configuration at the phosphorus atom. The (Rp).8-azolo-cAMPS products are analogues of cAMP that regulates a broad range of essential cellular functions.
Experimental
1H NMR spectra were recorded in CDCl3 or MeOH-d4 at 200 and 300 MHz. The 13C NMR spectra were recorded at 75 and 100 MHz. Chemical shifts are reported in ppm using CHCl3 (7.24 ppm) and CDCl3 (77 ppm) as references, and in MeOH-d4, 3.30 ppm in 1H NMR and 49.0 ppm in 13C NMR. The 31P spectra were recorded in CDCl3, MeOH-d4 at 81 MHz or 121 MHz with a Bruker DPX 200 or 300 instrument with 85% H3PO4 as an external reference. Mass spectra were recorded at 70 eV with a Fisons VG Prospectrometer. The spectra are presented as m/z (% relative intensity). Electrospray spectra were obtained with a Micromass QTOF 2 W spectrometer with electrospray ionisation quadrupole time of flight. Merck silica gel 60 (230–400) was used for flash chromatography.
(S p)-2′O-(tert-Butyldimethylsilyl)-8-(imidazol-1-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (2, C26H35N8O5PSi)
A solution of 143 mg imidazole (2.1 mmol) in 5 cm3 dry DMF was added slowly via a syringe to an oven-dried flask containing 84 mg powdered NaH (60% dispersion in mineral oil, 2.1 mmol) and 8 cm3 anhydrous DMF under argon. The mixture was stirred at room temperature for 30 min before a solution of 1.22 g (Sp)-8-bromoadenosine-2-O′-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (1, 2.0 mmol) in 8 cm3 DMF was added. The mixture was stirred at room temperature for 30 min and finally at 80 °C for 4 h. The solvent was removed at reduced pressure, and the residual material subjected to flash chromatography on silica gel using 10% methanol in CH2Cl2. Yield: 586 mg (49%) of a white solid material; 1H NMR (CDCl3, 300 MHz): δ = − 0.08 (3H, s, Si-Me), − 0.03 (3H, s, Si-Me), 0.71 (9H, s, Si-tBu), 3.84–3.88 (1H, m, NH), 4.11–4.18 (3H, m), 4.42–4.54 (3H, m), 5.19 (1H, d, J = 5.1 Hz), 5.43 (1H, s), 5.57–5.62 (1H, m), 6.05 (2H, br s, NH2), 7.26 (1H, s, H-imidazole), 7.27–7.31 (5H, m), 7.36 (1H, s, H-imidazole), 7.96 (1H, s, H-imidazole), 8.27 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = − 5.2, − 4.7, 18.0, 25.5, 45.3, 68.2, 71.3, 73.4, 76.3 (partly overlapped with CDCl3 peak), 93.0, 117.3, 119.65, 127.1, 127.6, 128.6, 131.1, 137.7, 138.6, 140.2, 149.5, 154.1, 155.45 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.46 ppm; HRMS (electrospray, TOF, ES+): m/z = 599.2313, calc. for C26H35N8O5PSi + H+ 599.2315.
(S p)-2′O-(tert-Butyldimethylsilyl)-8-(1,2,4-triazol-1-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (3, C25H34N9O5PSi)
A solution of 145 mg 1,2,4-triazole (2.1 mmol) in 5 cm3 dry DMF was added slowly via a syringe to an oven-dried flask containing 84 mg powdered NaH (60% dispersion in mineral oil, 2.1 mmol) and 8 cm3 anhydrous DMF under argon. The mixture was stirred at room temperature for 30 min before a solution of 1.22 g (Sp)-8-bromoadenosine-2′O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (1, 2.0 mmol) in 8 cm3 DMF was added. The mixture was stirred at room temperature for 30 min and finally at 85 °C overnight (20 h). The solvent was distilled off at reduced pressure, and the residual material subjected to flash chromatography on silica gel using 8% methanol in CH2Cl2 to afford 659 mg (55%) of the triazole product 3 as a white solid. 1H NMR (CDCl3, 300 MHz): δ = 0.02 (6H, s, 2 × Si-Me), 0.81 (9H, s, Si-tBu), 3.78–3.82 (1H, m, NH), 4.13–4.18 (3H, m), 4.40–4.59 (3H, m), 5.07 (1H, d, J = 5.1 Hz), 5.76–5.79 (1H, m), 6.06 (2H, br s, NH2), 6.56 (1H, s), 7.24–7.30 (5H, m), 8.17 (1H, s, H-triazole), 8.27 (1H, s, H-2), 8.88 (1H, s, H-triazole) ppm; 13C NMR (CDCl3, 75 MHz): δ = -5.2, -4.6, 18.1, 25.5, 45.4, 68.3, 71.4, 73.4, 76.3, 93.45, 117.3, 127.1, 127.55, 128.6, 138.65, 139.2, 145.4, 150.1, 153.6, 154.1, 155.4 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.53 ppm; HRMS (electrospray, TOF, ES+): m/z = 600.2263, calc. for C25H34N9O5PSi + H+ 600.2268.
(S p)-2′O-(tert-Butyldimethylsilyl)-8-(1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (4, C25H34N9O5PSi) and (S p)-2′O-(tert-butyldimethylsilyl)-8-(1,2,3-triazol-1-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (5, C25H34N9O5PSi)
A solution of 211 mg 1,2,3-triazole (3.06 mmol) in 5 cm3 dry DMF was added slowly via a syringe to an oven-dried flask containing 122 mg powdered NaH (60% dispersion in mineral oil, 3.06 mmol) and 10 cm3 anhydrous DMF under argon. The mixture was stirred at room temperature for 30 min before a solution of 1.70 g (Sp)-8-bromoadenosine-2'O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (1, 2.78 mmol) in 10 cm3 DMF was added. The mixture was stirred at room temperature for 30 min and finally at 85 °C overnight (20 h). The solvent was removed at reduced pressure, and the residual material subjected to flash chromatography on silica gel using 8% MeOH in CH2Cl2 to furnish the white product as a mixture (4 + 5), yield 880 mg (53%) in ratio 3:2 by 1H NMR spectroscopy analysis. The isomers were subjected twice to separation by flash chromatography using 8% methanol in CH2Cl2. Yields: 365 mg of isomer 4 and 210 mg of isomer 5.
4: 1H NMR (CDCl3, 300 MHz): δ = 0.00 (3H, s, Si-Me), 0.02 (3H, s, Si-Me), 0.80 (9H, s, Si-tBu), 3.52–3.56 (1H, m, NH), 4.14–4.21 (3H, m), 4.39–4.63 (3H, m), 5.04 (1H, d, J = 5.2 Hz), 5.80–5.85 (1H, m), 5.97 (2H, br s, NH2), 6.51 (1H, s), 7.24–7.32 (5H, m), 7.99 (2H, s, H-triazole), 8.35 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = − 5.25, − 4.6, 18.1, 25.5, 45.4, 68.4, 71.6, 73.6, 76.3, 93.8, 117.3, 127.1, 127.6, 128.6, 137.7, 138.8, 141.0, 149.9, 154.3, 155.6 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.47 ppm; HRMS (electrospray, TOF, ES+): m/z = 600.2263, calc. for C25H34N9O5PSi + H+ 600.2268.
5: 1H NMR (CDCl3, 200 MHz): δ = 0.025 (3H, s, Si-CH3), 0.04 (3H, s, Si-Me), 0.81 (9H, s, Si-tBu), 3.86–3.92 (1H, m, NH), 4.11–4.22 (3H, m), 4.35–4.57 (3H, m), 5.12 (1H, d, J = 5.1 Hz), 5.71–5.79 (1H, m), 6.11 (2H, br s, NH2), 6.48 (1H, s), 7.24–7.30 (5H, m), 7.86 (1H, d, J = 1 Hz, H-triazole), 8.26 (1H, s, H-2), 8.30 (1H, d, J = 1 Hz, H-triazole) ppm; 13C NMR (CDCl3, 75 MHz): δ = -5.15, -4.7, 18.1, 25.5, 45.4, 68.3, 71.4, 73.3, 76.3, 93.8, 117.5, 125.5, 127.1, 127.5, 128.6, 133.9, 138.7, 139.3, 150.1, 154.2, 155.65 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.58 ppm; HRMS (electrospray, TOF, ES+): m/z = 600.2285, calc. for C25H34N9O5PSi + H+ 600.2268.
(S P)-2′O-(tert-Butyldimethylsilyl)-8-(4-methyl-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (8, C26H36N9O5PSi)
A solution of 146 mg 4-methyl-1,2,3-triazole (1.75 mmol) in 5 cm3 dry DMF was added slowly via a syringe to an oven-dried flask containing 70 mg powdered NaH (60% dispersion in mineral oil, 1.75 mmol) and 5 cm3 anhydrous DMF under argon. The mixture was stirred for 30 min at room temperature before a solution of 915 mg (SP)-8-bromoadenosine-2'O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (1, 1.5 mmol) in 7 cm3 DMF was added. The mixture was stirred at room temperature for 30 min and then at 90 °C overnight (20 h). The solvent was removed at reduced pressure, and the residual material subjected to flash chromatography on silica gel using 8% methanol in CH2Cl2 to afford 380 mg (41%) of (8 + 10) as a mixture in the ratio 6:1 (1H NMR) as a white solid material. The products were separated and purified by flash chromatography twice. Yield of 8: 265 mg; 1H NMR (CDCl3, 200 MHz): δ = -0.02 (3H, s, Si-Me), 0.03 (3H, s, Si-Me), 0.82 (9H, s, Si-tBu), 2.46 (3H, s, Me), 3.36–3.46 (1H, m, NH), 4.13–4.21 (3H, m), 4.40–4.59 (3H, m), 5.05 (1H, d, J = 5.3 Hz), 5.75–5.85 (3H, m), 6.69 (1H, s), 7.24–7.32 (5H, m), 7.74 (1H, s, H-triazole), 8.35 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = − 5.25, -4.6, 10.8, 18.1, 25.5, 45.4, 68.5, 71.4, 73.6, 76.6 (partly overlapped with CDCl3 peak), 93.8, 117.3, 127.1, 127.6, 128.6, 137.6, 138.7, 141.3, 148.1, 150.0, 153.9, 155.3 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.88 ppm; HRMS (electrospray, TOF, ES+): m/z = 614.2418, calc. for C26H36N9O5PSi + H+ 614.2424.
(S P)-2′O-(tert-Butyldimethylsilyl)-8-(4-chloro-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (11, C25H33ClN9O5PSi)
A solution of 124 mg 4-chloro-1,2,3-triazole (1.2 mmol) in 5 cm3 dry DMF was added slowly via a syringe to an oven-dried flask containing 48 mg powdered NaH (60% dispersion in mineral oil, 1.2 mmol) and 3 cm3 anhydrous DMF under argon. The mixture was stirred at room temperature for 30 min before a solution of 611 mg (Sp)-8-bromoadenosine-2'O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (1, 1.0 mmol) in 5 cm3 DMF was added. The mixture was stirred at room temperature for 30 min and finally at 85 °C overnight (20 h). The solvent was removed at reduced pressure, and the residual material subjected to flash chromatography on silica gel using 5% MeOH in CH2Cl2 to afford 350 mg (55%) of (9 + 11) as a white solid mixture in the ratio 3:1 (1H NMR). Pure 9 was isolated after flash chromatography twice. Yield: 215 mg; 1H NMR (CDCl3, 300 MHz): δ = 0.03 (6H, s, 2 × Si-Me), 0.82 (9H, s, Si-tBu), 3.65–3.68 (1H, m, NH), 4.14–4.19 (3H, m), 4.38–4.57 (3H, m), 5.06 (1H, d, J = 5.1 Hz), 5.77–5.83 (1H, m), 6.05 (2H, br s, NH2), 6.45 (1H, s), 7.25–7.32 (5H, m), 7.87 (1H, s, H-triazole), 8.31 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = -5.2, -4.6, 18.1, 25.5, 45.4, 68.3, 71.5, 73.5, 76.2, 93.7, 117.2, 127.1, 127.6, 128.6, 136.3, 138.7, 140.3, 141.1, 149.9, 154.4, 155.6 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.97 ppm; HRMS (electrospray, TOF, ES+): m/z = 634.1887, calc. for C25H33ClN9O5PSi + H+ 634.1878.
(S p)-8-Azidoadenosine-2′O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (12, C23H32N9O5PSi)
A solution of 1.22 g (Sp)-8-bromoadenosine-2′O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (1, 2.0 mmol) in 20 cm3 DMF was stirred together with 390 mg NaN3 (6.0 mmol) at 60 °C for 20 h. The solvent was distilled off at reduced pressure, and the residual material subjected to flash chromatography on silica gel using 10% methanol. 1H NMR (CDCl3, 200 MHz): δ = 0.03 (3H, s, Si-Me), 0.04 (3H, s, Si-Me), 0.85 (9H, s, Si-tBu), 3.45–3.55 (1H, m, NH), 4.09–4.21 (3H, m), 4.40–4.56 (3H, m), 4.85 (1H, d, J = 5.1 Hz), 5.44–5.49 (1H, m), 5.50 (2H, br s, NH2), 5.79 (1H, s), 7.27–7.33 (5H, m), 8.20 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = -5.0, − 4.7, 18.1, 25.6, 45.4, 68.4, 71.1, 73.1, 76.6 (partly overlapped with CDCl3 peak), 91.8, 117.8, 127.1, 127.6, 128.6, 138.7, 144.8, 150.0, 152.6, 153.5 ppm; 31P NMR (CDCl3, 121 MHz): δ = 7.43 ppm; HRMS (electrospray, TOF, ES+): m/z = 574.2101, calc. for C23H32N9O5PSi + H+ 574.2111.
(R p)-8-(Imidazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (15, C13H13N7O5PS)
A 1.8 M solution of LDA in THF/heptane/ethylbenzene (0.37 cm3, 0.66 mmol) was added to a solution of 358 mg (SP)-2′O-(tert-butyldimethylsilyl)-8-(imidazol-1-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (2, 0.6 mmol) in 10 cm3 dry THF at − 78 °C. The mixture was stirred under argon at this temperature for 10 min before 0.10 cm3 CS2 (1.8 mmol) was added. The cooling bath was removed after 10 min. The reaction mixture was stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated (RP)-2′O-(tert-butyldimethylsilyl)-8-(imidazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid lithium salt (13) as a solid. The product was dried and dissolved in 10 cm3 MeOH and 310 mg NH4F (8.4 mmol) added to the solution. The mixture was stirred at 45 °C for 4 h, 166 mg nBu3N (0.9 mmol) added and the solvent was removed at reduced pressure. The residue was subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:nBu3N (90:10:1). A second flash chromatography operation using CH2Cl2:CH3OH:nBu3N (95:5:1) gave the ammonium salt 15, that contained some nBu3N. The crude product was purified by dissolution in CH2Cl2 and reprecipitation by addition of hexane. Yield: 160 mg (45% from 2); 1H NMR (CDCl3, 300 MHz): δ = 0.93 (9H, t, J = 7.3 Hz, 3 × Me), 1.34–1.42 (6H, m, 3 × CH2), 1.65–1.72 (6H, m, 3 × CH2), 2.99–3.04 (6H, m, 3 × CH2), 4.25–4.39 (3H, m), 5.19 (1H, d, J = 5.0 Hz), 5.35–5.47 (1H, m), 5.61 (1H, s), 6.04 (2H, br s, NH2), 7.19 (1H, s, H-imidazole), 7.44 (1H, s, H-imidazole), 8.10 (1H, s, H-imidazole), 8.19 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = 13.6, 20.1, 25.2, 51.9, 67.0, 71.2, 72.0, 76.2, 91.3, 117.4, 119.8, 130.7, 138.0, 140.8, 149.7, 153.25, 155.2 ppm; 31P NMR (CDCl3, 121 MHz): δ = 56.39 ppm; HRMS (electrospray, TOF, ES−): m/z = 410.0422, calc. for C13H13N7O5PS 410.0436.
(R p)-8-(1,2,4-Triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (16, C12H12N8O5PS−)
A 1.8 M solution of LDA in THF/heptane/ethylbenzene (0.57 cm3, 1.02 mmol) was added to a solution of 560 mg (SP)-2′O-(tert-butyldimethylsilyl)-8-(1,2,4-triazol-1-yl)adenosine-3',5'-cyclic N-benzylphosphoramidate (3, 0.93 mmol) in 15 cm3 dry THF at − 40 °C. The mixture was stirred under argon at this temperature for 10 min before 0.17 cm3 CS2 (2.79 mmol) was added. The cooling bath was removed after 10 min and the reaction mixture stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated the solid (RP)-2′O-(tert-butyldimethylsilyl)-8-(1,2,4-triazol-1-yl)adenosine-3',5'-cyclic thiophosphoric acid lithium salt (14). The dried salt was dissolved in 15 cm3 methanol, 482 mg NH4F (13.0 mmol) added, and the mixture stirred at 45 °C for 4 h. nBu3N (258 mg, 1.4 mmol) was added to the solution and the solvent was removed at reduced pressure. The residue was subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:nBu3N (90:10:1). Repeated flash chromatography using CH2Cl2:CH3OH:n-Bu3N (93:7:1) gave the ammonium salt 16, that contained some nBu3N. The product was purified by dissolution in CH2Cl2 and reprecipitation by hexane addition. Yield: 255 mg (46% from 3); 1H NMR (CDCl3, 300 MHz): δ = 0.94 (9H, t, J = 7.3 Hz, 3 × Me), 1.34–1.42 (6H, m, 3 × CH2), 1.69–1.74 (6H, m, 3 × CH2), 2.99–3.04 (6H, m, 3 × CH2), 4.29–4.43 (3H, m), 5.06 (1H, d, J = 5.2 Hz), 5.52–5.57 (1H, m), 5.87 (2H, br s, NH2), 6.43 (1H, s), 8.21 (1H, s, H-triazole), 8.26 (1H, s, H-2), 8.89 (1H, s, H-triazole) ppm; 13C NMR (CDCl3, 75 MHz): δ = 13.6, 20.1, 25.2, 51.9, 67.0, 71.9, 72.0, 76.2, 91.7, 117.3, 139.6, 145.5, 150.2, 153.5, 153.8, 155.2 ppm; 31P NMR (CDCl3, 121 MHz): δ = 56.69 ppm; HRMS (electrospray, TOF, ES−): m/z = 411.0391, calc. for C12H12N8O5PS− 411.0389.
(R p)-8-(1,2,3-Triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (20, C12H12N8O5PS−)
A 1.8 M solution of LDA in THF/heptane/ethylbenzene (0.3 cm3, 0.55 mmol) was added to a solution of 300 mg (SP)-2’O-(tert-butyldimethylsilyl)-8-(1,2,3-triazol-2-yl)adenosine-3',5'-cyclic N-benzylphosphoramidate (4, 0.5 mmol) in 10 cm3 dry THF at − 50 °C. The mixture was stirred under argon at this temperature for 10 min before 0.09 cm3 CS2 (1.5 mmol) was added. The cooling bath was removed after 10 min and the reaction mixture stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated a solid that was filtered off. The product was (RP)-2′O-(tert-butyldimethylsilyl)-8-(1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid lithium salt (17). The solid product was dried, dissolved in 5 cm3 methanol and 259 mg NH4F (7 mmol) was added. The mixture was stirred at 45 °C for 4 h before 140 mg nBu3N (0.75 mmol) was added. The solvent was removed at reduced pressure and the residual material subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:nBu3N (90:10:1). The product was the ammonium salt 20 that contained some nBu3N. The product was purified by dissolution in CH2Cl2 and reprecipitation by addition of hexane. Yield: 125 mg (42% from 4); 1H NMR (CDCl3, 300 MHz): δ = 0.94 (9H, t, J = 7.3 Hz, 3 × Me), 1.35–1.42 (6H, m, 3 × CH2), 1.69–1.73 (6H, m, 3 × CH2), 2.99–3.04 (6H, m, 3 × CH2), 4.30–4.45 (3H, m), 5.06 (1H, d, J = 5.2 Hz), 5.59–5.65 (1H, m), 5.99 (2H, br s, NH2), 6.47 (1H, s), 8.00 (2H, s, H-triazole), 8.27 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = 13.6, 20.1, 25.2, 51.9, 67.1, 72.1, 72.3, 76.1, 92.0, 117.3, 137.9, 141.4, 149.9, 153.5, 155.4 ppm; 31P NMR (CDCl3, 121 MHz): δ = 56.08 ppm; HRMS (electrospray, TOF, ES−): m/z = 411.0379, calc. for C12H12N8O5PS− 411.0389.
(R p)-8-(4-Methyl-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic phosphoric acid tri-n-butylammonium salt (21, C13H14N8O5PS−)
A 1.8 M solution of LDA in THF/heptane/ethylbenzene (0.21 cm3, 0.38 mmol) was added to a solution of 215 mg (SP)-2'O-(tert-butyldimethylsilyl)-8-(4-methyl-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic N-benzylphosphoramidate (8, 0.35 mmol) in 7 cm3 dry THF at − 50 °C. The mixture was stirred under argon at this temperature for 10 min before 0.07 cm3 CS2 (1.05 mmol) was added. The cooling bath was removed after 10 min, the mixture stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated the solid (RP)-2′O-(tert-butyldimethylsilyl)-8-(4-methyl-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid lithium salt (18). The product was dried, dissolved in 4 cm3 MeOH and 181 mg NH4F (4.9 mmol) added. The mixture was stirred at 45 °C for 4 h, 97 mg nBu3N (0.52 mmol) added and the solvent was removed at reduced pressure. The residue was subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:nBu3N (90:10:1). A second flash chromatography operation using CH2Cl2:CH3OH:nBu3N (93:7:1) gave the ammonium salt 23, that contained some nBu3N. The product was purified by dissolution in CH2Cl2 and reprecipitation after hexane addition. Yield: 84 mg (39% from 8); 1H NMR (CDCl3, 200 MHz): δ = 0.92 (9H, t, J = 7.3 Hz, 3 × CH3), 1.32–1.40 (6H, m, 3 × CH2), 1.61–1.65 (6H, m, 3 × CH2), 2.44 (3H, s, Me), 2.82–2.90 (6H, m, 3 × CH2), 4.34–4.41 (3H, m), 5.03 (1H, d, J = 5.2 Hz), 5.59–5.65 (1H, m), 5.86 (2H, br s, NH2), 6.54 (1H, s), 7.75 (1H, s, H-triazole), 8.24 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = 10.9, 13.6, 20.1, 25.2, 51.9, 67.1, 72.3, 72.4, 76.2, 92.0, 117.3, 137.9, 141.7, 148.1, 149.9, 153.5, 155.4 ppm; 31P NMR (CDCl3, 121 MHz): δ = 56.61 ppm; HRMS (electrospray, TOF, ES−): m/z = 4,250,539, calc. for C13H14N8O5PS− 425.0545.
(R p)-8-(4-Chloro-1,2,3-triazol)-2-yl)adenosine-3',5'-cyclic thiophosphoric acid tri-n-butylammonium salt (22, C12H11ClN8O5PS−)
A 1.8 M solution of LDA in THF/heptane/ethylbenzene (0.21 cm3, 0.38 mmol) was added to a solution of 220 mg (SP)-2'O-(tert-butyldimethylsilyl)-8-(4-chloro-1,2,3-triazol-2-yl)adenosine-3',5'-cyclic N-benzylphosphoramidate (9, 0.35 mmol) in 7 cm3 dry THF at − 50 °C. The mixture was stirred under argon at this temperature for 10 min before 0.07 cm3 CS2 (1.05 mmol) was added. The reaction mixture was stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated the solid (RP)-2′O-(tert-butyldimethylsilyl)-8-(4-chloro-1,2,3-triazol-2-yl)adenosine-3',5'-cyclic thiophosphoric acid lithium salt (19). The product was dried, dissolved in 4 cm3 methanol and 181 mg NH4F (4.9 mmol) added. The mixture was stirred at 45 °C for 4 h, 97 mg nBu3N (0.52 mmol) added and the solvent was removed at reduced pressure. The residue was subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:n-Bu3N (90:10:1). A second flash chromatography operation using CH2Cl2:CH3OH:n-Bu3N (93:7:1) gave the ammonium salt 22, which contained some n-Bu3N. The product was purified by dissolution in CH2Cl2 and reprecipitation after hexane addition. Yield: 92 mg (41% from 9); 1H NMR (CDCl3, 300 MHz): δ = 0.94 (9H, t, J = 7.3 Hz, 3 × Me), 1.34–1.42 (6H, m, 3 × CH2), 1.65–1.72 (6H, m, 3 × CH2), 2.94–3.03 (6H, m, 3 × CH2), 4.30–4.40 (3H, m), 5.05 (1H, d, J = 5.2 Hz), 5.54–5.59 (1H, m), 5.94 (2H, br s, NH2), 6.38 (1H, s), 7.89 (1H, s, H-triazole), 8.27 (1H, s, H-2) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 13.6, 20.1, 25.2, 51.9, 67.2, 72.1, 72.2, 76.2, 92.0, 117.2, 136.5, 140.7, 141.1, 149.9, 153.7, 155.4 ppm; 31P NMR (CDCl3, 121 MHz): δ = 56.83 ppm; HRMS (electrospray, TOF, ES−): m/z = 444.9990, calc. for C12H11ClN8O5PS− 444.9999.
(R p)-8-(1,2,3-Triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (23, C12H12N8O5PS−)
A 1.8 M solution of LDA in THF/heptane/ethylbenzene (0.2 cm3, 0.34 mmol) was added to a solution of 180 mg (SP)-2′O-(tert-butyldimethylsilyl)-8-(1,2,3-triazol-1-yl)adenosine-3′',5′-cyclic N-benzylphosphoramidate (5, 0.3 mmol) in 7 cm3 dry THF at − 50 °C. The mixture was stirred under argon at this temperature for 10 min before 0.06 cm3 CS2 (0.9 mmol) was added. The reaction mixture was stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated the solid (RP)-2′O-(tert-butyldimethylsilyl)-8-(1,2,3-triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid lithium salt. The latter was dried, dissolved in 4 cm3 MeOH and 155 mg NH4F (4.2 mmol) was added. The mixture was stirred at 45 °C for 4 h, 85 mg n-Bu3N (0.45 mmol) was added, the solvent removed at reduced pressure and the residue subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:nBu3N (90:10:1). The flash chromatography was repeated using CH2Cl2:CH3OH:nBu3N (93:7:1). The ammonium salt 23 thus obtained contained some n-Bu3N and was further purified by dissolution in CH2Cl2 and reprecipitation by hexane addition. Yield: 70 mg (40% from 5); 1H NMR (CDCl3, 300 MHz): δ = 0.94 (9H, t, J = 7.3 Hz, 3 × Me), 1.34–1.42 (6H, m, 3 × CH2), 1.69–1.74 (6H, m, 3 × CH2), 2.99–3.04 (6H, m, 3 × CH2), 4.30–4.41 (3H, m), 5.11 (1H, d, J = 5.2 Hz), 5.46–5.50 (1H, m), 5.87 (2H, br s, NH2), 6.29 (1H, s), 7.88 (1H, d, J = 1 Hz, H-triazole), 8.28 (1H, s, H-2), 8.30 (1H, d, J = 1 Hz, H-triazole) ppm; 13C NMR (CDCl3, 75 MHz): δ = 13.6, 20.1, 25.2, 51.9, 67.0, 71.9, 72.1, 76.2, 92.0, 117.5, 125.8, 133.9 137.9, 149.1, 154.3, 155.6 ppm; 31P NMR(CDCl3, 121 MHz): δ = 56.00 ppm; HRMS (electrospray, TOF, ES−): m/z = 411.0382, calc. for C12H12N8O5PS− 411.0389.
(R p)-8-Azidoadenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (24, C10H10N8O5PS−)
A 1.8 M Solution of LDA in THF/heptane/ethylbenzene (0.61 cm3, 1.1 mmol) was added to a solution of 575 mg (SP)-8-azidoadenosine-2′O-(tert-butyldimethylsilyl)-3′,5′-cyclic N-benzylphosphoramidate (12, 1.0 mmol) in 15 cm3 dry THF at − 78 °C. The mixture was stirred under argon at this temperature for 10 min before 0.18 cm3 CS2 (3.0 mmol) was added. The reaction mixture was stirred at room temperature for 3 h before the solvent was partially removed at reduced pressure. Addition of hexane to the residual solution precipitated a solid that was filtered off. The resulting (RP)-8-azidoadenosine-2′O-(tert-butyldimethylsilyl)-3′,5′-cyclic thiophosphoric acid lithium salt was dried and dissolved in 10 cm3 methanol and 476 mg NH4F (14.0 mmol) added. The mixture was stirred at 45 °C for 4 h, 277 mg nBu3N (1.5 mmol) added and the solvent removed at reduced pressure. The residue was subjected to flash chromatography on silica gel using CH2Cl2:CH3OH:nBu3N (90:10:1). Repetitive flash chromatography using CH2Cl2:CH3OH:n-Bu3N (93:7:1) gave the ammonium salt 24, that contained some nBu3N. The product was purified by dissolution in CH2Cl2 and reprecipitation by addition of hexane. Yield: 273 mg (48% from 12); 1H NMR (CDCl3, 200 MHz): δ = 0.94 (9H, t, J = 7.3 Hz, 3 × CH3), 1.33–1.44 (6H, m, 3 × CH2), 1.63–1.79 (6H, m, 3 × CH2), 2.99–3.04 (6H, m, 3 × CH2), 4.30–4.41 (3H, m), 4.89 (1H, d, J = 5.0 Hz), 5.27–5.33 (1H, m), 5.58 (2H, br s, NH2), 5.84 (1H, s), 8.14 (1H, s, H-2) ppm; 13C NMR (CDCl3, 75 MHz): δ = 13.6, 20.1, 25.2, 51.9, 67.1, 71.6, 71.7, 76.2, 90.0, 117.8, 145.2, 150.0, 152.0, 153.4 ppm; 31P NMR (CDCl3, 121 MHz): δ = 56.72 ppm; HRMS (electrospray, TOF, ES−): m/z = 385.0245, calc. for C10H10N8O5PS− 385.0232.
(R p)-8-(Imidazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid sodium salt (25, C13H13N7O5PS−)
(Rp)-8-(Imidazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (15, 130 mg, 0.21 mmol) was dissolved in 0.1 M NaOH in MeOH (2.1 cm3). The sodium salt was precipitated by addition of diethyl ether. The solvent was decanted and the precipitate washed with diethyl ether. The ether was removed and the white solid sodium salt dried under vacuum. Yield: 81 mg (88%); 1H NMR (MeOH-d4, 300 MHz): δ = 4.22–4.35 (3H, m), 5.14 (1H, d, J = 5.2 Hz), 5.35–5.42 (1H, m), 5.51 (1H, s), 7.26 (1H, s, H-imidazole), 7.66 (1H, s, H-imidazole), 8.20 (1H, s, H-imidazole), 8.25 (1H, s, H-2) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 68.3, 72.5, 73.4, 77.5, 93.1, 118.3, 121.9, 130.7, 139.65, 141.7, 150.6, 154.75, 157.3 ppm; 31P NMR (MeOH-d4, 121 MHz): δ = 57.92 ppm; HRMS (electrospray, TOF, ES−): m/z = 410.0425, calc. for C13H13N7O5PS− 410.0436.
(R p)-8-(1,2,4-Triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid sodium salt (26, C12H12N8O5PS−)
Product 26 was prepared as above from (RP)-8-(1,2,4-triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (16) in 0.1 M NaOH in MeOH. Yield: 92%; 1H NMR (MeOH-d4, 200 MHz): δ = 4.12–4.35 (3H, m), 5.03 (1H, d, J = 5.3 Hz), 5.45–5.52 (1H, m), 6.27 (1H, s), 8.26 (1H, s, H-triazole), 8.34 (1H, s, H-2), 9.16 (1H, s, H-triazole) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 68.3, 73.1, 73.3, 77.4, 93.55, 118.3, 140.7, 147.6, 151.3, 154.4, 154.9, 157.5 ppm; 31P NMR (MeOH-d4, 121 MHz): δ = 58.06 ppm; HRMS (electrospray, TOF, ES−): m/z = 411.0390, calc. for C12H12N8O5PS− 411.0389.
(R p)-8-(1,2,3-Triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid sodium salt (27, C12H12N8O5PS−)
Product 27 was prepared as above from (RP)-8-(1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (20) in 0.1 M NaOH in MeOH. Yield: 90%; 1H NMR (MeOH-d4, 300 MHz): δ = 4.16–4.40 (3H, m), 5.03 (1H, d, J = 5.3 Hz), 5.41–5.49 (1H, m), 6.30 (1H, s), 8.18 (2H, s, H-triazole), 8.28 (1H, s, H-2) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 68.3, 73.2, 73.3, 77.3, 93.9, 118.0, 139.3, 142.3, 151.1, 155.1, 157.5 ppm; 31P NMR (MeOH-d4, 121 MHz): δ = 57.47 ppm; HRMS (electrospray, TOF, ES−): m/z = 411.0386, calc. for C12H12N8O5PS− 411.0389.
(R p)-8-(4-Methyl-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic phosphoric acid sodium salt (28, C13H14N8O5PS−)
Product 28 was prepared as above from (RP)-8-(4-methyl-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (21) in 0.1 M NaOH in MeOH. Yield: 91%; 1H NMR (MeOH-d4, 200 MHz): δ = 2.46 (3H, s, CH3), 4.14–4.34 (3H, m), 5.02 (1H, d, J = 5.4 Hz), 5.41–5.50 (1H, m), 6.39 (1H, s), 7.96 (1H, s, H-triazole), 8.26 (1H, s, H-2) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 10.6, 68.3, 73.2, 73.3, 77.3, 93.9, 117.95, 139.1, 142.6, 149.7, 151.0, 154.8, 157.3 ppm; 31P NMR (MeOH-d4, 121 MHz): δ = 57.95 ppm; HRMS (electrospray, TOF, ES−): m/z = 425.0533, calc. for C13H14N8O5PS− 425.0545.
(R p)-8-(4-Chloro-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid sodium salt (29, C12H11ClN8O5PS−)
Product 29 was prepared as above from (RP)-8-(4-chloro-1,2,3-triazol-2-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (22) in 0.1 M NaOH in MeOH. Yield: 87%; 1H NMR (MeOH-d4, 300 MHz): δ = 4.16–4.35 (3H, m), 5.04 (1H, d, J = 5.3 Hz), 5.43–5.48 (1H, m), 6.20 (1H, s), 8.22 (1H, s, H-triazole), 8.28 (1H, s, H-2) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 68.3, 73.2, 73.4, 77.3, 93.8, 118.0, 137.8, 141.7, 142.1, 151.0, 155.25, 157.5 ppm; 31P NMR (MeOH-d4, 121 MHz): δ = 57.31 ppm; HRMS (electrospray, TOF, ES−): m/z = 444.9991, calc. for C12H11ClN8O5PS− 444.9999.
(R p)-8-(1,2,3-Triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid sodium salt (30, C12H12N8O5PS−)
Product 30 was prepared as above from (RP)-8-(1,2,3-triazol-1-yl)adenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (23) in 0.1 M NaOH in MeOH. Yield: 92%; 31P NMR (MeOH-d4, 121 MHz): δ = 57.3 ppm; HRMS (electrospray, TOF, ES−): m/z = 411.0380, calc. for C12H12N8O5PS− 411.0389.
(R p)-8-Azidoadenosine-3′,5′-cyclic thiophosphoric acid sodium salt (31, C12H12N8O5PS−)
Product 31 was prepared as above from (RP)-8-azidoadenosine-3′,5′-cyclic thiophosphoric acid tri-n-butylammonium salt (24) in 0.1 M NaOH in MeOH. Yield: 87%; 1H NMR (MeOH-d4, 300 MHz): δ = 4.15–4.33 (3H, m), 4.78 (1H, d, J = 5.3 Hz), 5.22–5.28 (1H, m), 5.84 (1H, s), 8.12 (1H, s, H-2) ppm; 13C NMR (MeOH-d4, 75 MHz): δ = 68.4, 73.1, 73.2, 77.5, 91.7, 118.4, 146.4, 151.1, 153.2, 155.6 ppm; 31P NMR (MeOH-d4, 121 MHz): δ = 58.26 ppm; HRMS (electrospray, TOF, ES−): m/z = 385.0224, calc. for C10H10N8O5PS− 385.0232.
References
Undheim K (2022) Eur J Med Chem Rep 5:5100037
Andrei M, Bjørnstad V, Langli G, Rømming C, Klaveness J, Tasken K, Undheim K (2007) Org Biomol Chem 5:2070
Weissinger EM, Oetrich K, Evans C, Genieser H-G, Schwede F, Dangers M, Dammann E, Kolb H-J, Mischak H, Ganser A, Kolch W (2004) Br J Cancer 91:186
Undheim K, Andrei M (2008) Process for The Preparation of an (Rp)-8-Substituted adenosine cyclic monophosphorothioate (cAMPS). Chem Abstr 148:331962
Genieser H-G, Schwede F, Rentsch A (2018) New Equatorially Modified Polymer Linked Multimers of Guanosine-3′,5’-Cyclic Monophosphates. Chem Abstr. 168:167813
Ahmed KA, Zhang T, Ono K, Tsutsuki H, Ida T, Akashi S, Miyata K, Oike Y, Akaike T, Sawa T (2017) Biol Pharm Bull 40:365
Undheim K (2002) Heteroaromatics via Palladium-Catalyzed Cross-Coupling. In: Negishi E (ed) Organopalladium Chemistry for Organic Synthesis. John Wiley Interscience, New York, p 409
Hartwig JF (2008) Acc Chem Res 41:1534
Taskén K, Aandal EM (2004) Physiol Rev 84:137
Francis SH, Blount MA, Zoraghi R, Corbin JD (2005) Front Biosci 10:2097
Holzer W, Ruso K (1992) J Heterocycl Chem 29:1203
Baraniak J, Stec WJ (1987) J Chem Soc Perkin Trans 1:1645
Undheim K, Taskén K, Klaveness J, Langli G, Bjørnstad V (2005) Purine Nucleotide Derivatives. Chem Abstr 167:307382
Funding
Open access funding provided by University of Oslo (incl Oslo University Hospital).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andrei, M., Undheim, K. Azolo substitution into the purine scaffold in nucleoside cyclic 3',5'-phosphorothioates. Monatsh Chem 153, 1213–1223 (2022). https://doi.org/10.1007/s00706-022-02980-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-02980-2