
Abstract
The caesium phosphates Cs3(H1.5PO4)2(H2O)2 and Cs3(H1.5PO4)2 were obtained from aqueous solutions, and Cs4P2O7(H2O)4 and CsPO3 from solid state reactions, respectively. Cs3(H1.5PO4)2, Cs4P2O7(H2O)4, and CsPO3 were fully structurally characterized for the first time on basis of single-crystal X-ray diffraction data recorded at − 173 °C. Monoclinic Cs3(H1.5PO4)2 (Z = 2, C2/m) represents a new structure type and comprises hydrogen phosphate groups involved in the formation of a strong non-symmetrical hydrogen bond (accompanied by a disordered H atom over a twofold rotation axis) and a very strong symmetric hydrogen bond (with the H atom situated on an inversion centre) with symmetry-related neighbouring anions. Triclinic Cs4P2O7(H2O)4 (Z = 2, P\(\bar{1}\)) crystallizes also in a new structure type and is represented by a diphosphate group with a P–O–P bridging angle of 128.5°. Although H atoms of the water molecules were not modelled, O···O distances point to hydrogen bonds of medium strengths in the crystal structure. CsPO3 is monoclinic (Z = 4, P21/n) and belongs to the family of catena-polyphosphates (MPO3)n with a repetition period of 2. It is isotypic with the room-temperature modification of RbPO3. The crystal structure of Cs3(H1.5PO4)2(H2O)2 was re-evaluated on the basis of single-crystal X-ray diffraction data at − 173 °C, revealing that two adjacent hydrogen phosphate anions are connected by a very strong and non-symmetrical hydrogen bond, in contrast to the previously described symmetrical bonding situation derived from room temperature X-ray diffraction data. In the four title crystal structures, coordination numbers of the caesium cations range from 7 to 12.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The recent interest in the family of caesium phosphates is mainly connected with the high proton conductivity of Cs(H2PO4) to be utilized as a potential electrolyte for intermediate temperature fuel cells [1,2,3] or for water electrolysis [4]. Another motivation to search for new caesium phosphates is related to acidic salts with formulae MxHy(AO4)z (M = Cs, Rb, K, Na, Li, NH4; A = S, Se, As, P) that likewise exhibit proton conductivity or have ferroelectric properties. In this context, the new proton conductor Cs3(H2PO4)(HPO4)·2H2O (= Cs3(H1.5PO4)2(H2O)2) has been characterized lately [5].
Numerous caesium phosphates have been completely structurally characterized so far, viz. the low-temperature form of Cs3(PO4) [6], Cs2(HPO4)(H2O)2 [7], Cs(H2PO4) in its low- and room-temperature forms [8,9,10], as a superprotonic conductor phase [11] and in its high-pressure forms [12], CsH5(PO4)2 [13], Cs(H3P2O7)(H2O) [14], Cs2(H2P2O7) [15], Cs6(P6O18)(H2O)6 [16], Cs3(P3O9)(H2O) [17], Cs4(P4O12)(H2O)4 [18], and Cs8(P8O24)(H2O)8 [19].
Here, we report on the crystal structure refinements of the four caesium phosphates Cs3(H1.5PO4)2(H2O)2, Cs3(H1.5PO4)2, Cs4P2O7(H2O)4, and CsPO3. On the basis of high-quality single-crystal X-ray data sets recorded at − 173 °C, it was possible to get full structural details for Cs3(H1.5PO4)2 and CsPO3. Up to now, for these compounds only X-ray powder data were available from previous studies [5, 20]. In addition, the hydrogen-bonding scheme in the crystal structure of Cs3(H1.5PO4)2(H2O)2 [5] was re-evaluated, and the hydrous diphosphate Cs4P2O7(H2O)4 is reported here for the first time.
Results and discussion
Results of bond valence sum (BVS) calculations [21] using the parameters provided by Brese and O’Keeffe [22] reveal values for CsFootnote 1 and P atoms in all structures very close to the expected formal total valencies of + I and + V, respectively, with the highest deviation being 0.15 valence units for some P atoms (Table 1).
Cs3(H1.5PO4)2(H2O)2
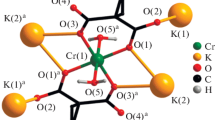
Concerning the previous single-crystal X-ray study of Cs3(H1.5PO4)2(H2O)2 at room temperature [5], the results of the current low-temperature study have a higher precision. However, the principal structural arrangement is the same with respect to the two refinements/models. Differences in bond lengths and angles for individual structure units between the two models are negligible and might be caused by different measurement temperatures. Selected bond lengths and angles resulting from the current refinement are collated in Table 1. Since the crystal structure of Cs3(H1.5PO4)2(H2O)2 has been discussed in detail, here only the main features are given. The crystal structure is built up of hydrogen phosphate tetrahedra connected through strong hydrogen bonds involving the hydrogen atoms H1 and H2 (Table 2) into undulating layers parallel (001). Under further contribution of two hydrogen-bonding interactions of medium strengths involving the water molecule (OW), a three-dimensional network is formed (Fig. 1). The two independent caesium cations are located in the voids of this arrangement and are bonded to eight (Cs1) and twelve (Cs2) O atoms (Fig. 2, Table 1).

The hydrogen-bonding network (green and yellow lines) of Cs3(H1.5PO4)2(H2O)2 in a projection along [010] involving the hydrogen phosphate tetrahedra and water molecules. Displacement ellipsoids are drawn at the 90% probability level; phosphate tetrahedra are red, O atoms are white, O atoms of OH groups are green, water O atoms are yellow, and H atoms are given as grey spheres of arbitrary radius (colour figure online)

The crystal structure of Cs3(H1.5PO4)2(H2O)2 in a projection along [100]. Displacement ellipsoids and colour codes are as in Fig. 1; Cs1 is given in dark blue, Cs2 in turquoise
The chief difference between the two models pertains to the very strong hydrogen bond developed between two phosphate tetrahedra involving O2 and its symmetry-related counterpart (O2···O2(− x + 1, − y, − z)) at a distance of ≈ 2.44 Å. In the previous room-temperature model [5], this hydrogen bond was suggested as being symmetric, with the H atom exactly positioned between the two O2 atoms at an inversion centre of space group Pbca (Wyckoff position 1a). Based on difference Fourier maps obtained from the current data set (Fig. 3), which clearly revealed two symmetry-related maxima in the vicinity of the inversion centre, we modelled the corresponding H atom (H2) as being statistically disordered, resulting in a non-symmetrical O2–H···O2′ hydrogen bond (Table 2). The bonding situation regarding such a very strong hydrogen bond between two hydrogen phosphate groups with disordered hydrogen atoms is similar as in other structures comprising tetrahedral oxoanions with OH groups, e.g. in (NH4)H5(PO4)2 [23], TlIH5(AsO4)2 [24], or Na5H3(SeO4)4(H2O)2 [25].

a Difference Fourier map around symmetry-related O2 atoms (red points) on basis of a model with omission of the respective H atoms; the two maxima related to electron densities of the disordered H atoms are discernible. b Fobs map with H atoms (pale pink dots). The sections are centred on the inversion centre at (½, 0, 0) and pass through the O2 atom. The width and height of the plots show 3 Å. Contours (solid: positive, dotted: negative, dashed: zero) are drawn at multiples of a 0.05 e−/Å3 and b 0.5 e−/Å3 (colour figure online)
Although the present low-temperature X-ray diffraction data for Cs3(H1.5PO4)2(H2O)2 clearly point to an unsymmetrical hydrogen bond O2–H2···O2(− x + 1, − y, − z) with a disordered H2 atom, it remains unclear whether this model also applies at room temperature, or whether the reported model [5] with a symmetrical hydrogen bond O2—H2—O2′ and with H2 situated at an inversion centre is correct at this temperature. Note that the O2···O2′ distance derived from the room-temperature measurement (2.445(7) Å [5]) is slightly longer than in the current low-temperature measurement (2.4343(19) Å), indicating an expansion of the structure. Therefore, the likelihood of a symmetric hydrogen bond is expected to decrease with higher temperature. In the end, this question (unsymmetrical versus symmetrical hydrogen bond) can be answered without ambiguity only on basis of temperature-dependent neutron diffraction data.
Cs3(H1.5PO4)2
Cs3(H1.5PO4)2 was reported to exist as a dehydration product of Cs3(H1.5PO4)2(H2O)2 and to be stable between ~ 50 and 275 °C. Except for unit cell parameters and space group assignment on basis of a laboratory X-ray powder study, no further structural details were given for this phase [5]. The unit cell parameters determined from polycrystalline Cs3(H1.5PO4)2 at 160 °C (a = 11.1693(4), b = 6.4682(2), c = 7.7442(3) Å, β = 71.822(2)°, V = 531.56 Å3) [5] are in good agreement with the values from the current single-crystal X-ray data at − 173 °C (Table 3). However, the space group derived from the powder study was reported to be C2 (No. 5), whereas the current refinement clearly indicates the higher C2/m space group symmetry.
The unique crystal structure of Cs3(H1.5PO4)2 at − 173 °C resembles that of the corresponding dihydrate described above and comprises two Cs, one P, three O, and two H atoms in the asymmetric unit. Cs1 is situated on Wyckoff position 2a (site symmetry 2/m), Cs2, P1, O1, O2, O3 are all situated on position 4i (m), H1 (8j; 1) is disordered about a twofold rotation axis, and H3 is situated on Wyckoff position 2b (2/m).
The P–O bond lengths distribution is typical for a hydrogen phosphate anion, with the P–OH bonds (involving O3 and O1) considerably longer (average 1.562 Å) than the P–O2 bond of 1.503 Å. Whereas the latter oxygen atom is not engaged in hydrogen bonding, each of the OH groups is involved in very strong to strong hydrogen bonding with its symmetry-related counterpart generated through a 2/m operation and a twofold rotation axis, respectively. Individual phosphate tetrahedra are connected through a very strong (O···O = 2.458(2) Å) and symmetrical hydrogen bond O3–H3–O3(− x + 1, y, − z) (Table 2) with H3 at a 2/m position to form a dimer with composition {H(HPO4)2}3−. Adjacent dimers are linked through additional strong hydrogen bonds (O···O = 2.55 Å) involving the disordered H1 atom (O1–H1···O1(− x + 1, y, − z − 1) into zig-zag [001] chains situated at x ≈ 0, y ≈ 0.5 (Fig. 4).

Hydrogen-bonded chains of hydrogen tetrahedra in the crystal structure of Cs3(H1.5PO4)2, viewed along [010]. Displacement ellipsoids and colour codes are as in Fig. 1
In comparison with hydrated Cs3(H1.5PO4)2(H2O)2 where the very short hydrogen bond is unsymmetrical and associated with disorder of the hydrogen atom, the bonding situation of the hydrogen phosphate tetrahedron is different in Cs3(H1.5PO4)2. As noted above, the hydrogen bond is symmetrical as revealed by difference Fourier maps, which clearly show a maximum at the inversion centre in-between the two symmetry-related oxygen atoms O1 (Fig. 5). However, it has to be noted that the electron density about hydrogen atoms is diffuse. Thus, even for a perfectly determined electron density, an H atom disordered about the 2/m position may result in a single maximum, if the disordered atoms are close to said position. In consequence, as previously, only neutron diffraction studies can unambiguously demonstrate the correctness of this model, since neutrons diffract at the nucleus.

a Difference Fourier map around symmetry-related O3 atoms (red points) on basis of a model with omission of the respective H atoms, showing one maximum situated at the 2/m position related to electron density of an ordered H atom; b Fobs map with H atom (pale pink dot). The plots represent y = 0 sections centred around (½, 0, 0) and show 3 Å in the x- and z-directions. Contours (solid: positive, dotted: negative, dashed: zero) are drawn at multiples of a 0.05 e−/Å3 and b 0.5 e−/Å3 (colour figure online)
The two caesium cations are situated between the hydrogen-bonded chains (Fig. 6). Each Cs1 symmetrically links four chains; it exhibits a coordination number of 10 in form of a distorted hexadecahedron [26]. Cs1 is bonded to two (non-H atom bearing) O3 atoms of two neighbouring chains with the shortest Cs–O bonds (3.07 Å) observed for this polyhedron, to four (disordered H atom bearing) symmetrically related O1 atoms at longer distances (3.22 Å), and to another four (H atom bearing) O3 atoms at the longest distance (3.41 Å). Each Cs2 likewise links four chains and is surrounded by eight O atoms in the shape of a distorted hexagonal bipyramid. The two shortly bonded O atoms O3 and O2 (d(Cs–O) ≈ 3.05 Å) define the axial O atoms, and the symmetrically related three pairs of O1 and O2 atoms define the six equatorial atoms with bond lengths ranging from 3.16 to 3.30 Å (Table 1).

The crystal structure of Cs3(H1.5PO4)2 in a projection along [001]. Displacement ellipsoids and colour codes are as in Fig. 2
Cs4P2O7(H2O)4
In the crystal structure of Cs4P2O7(H2O)4 (Fig. 7), isolated diphosphate anions are organised in layers parallel (010), thereby sandwiching adjacent layers composed of caesium cations (Cs3, Cs4) and the four water molecules along the [010] direction. The remaining two caesium cations, Cs1 and Cs2, are situated in-between individual diphosphate groups in the anionic layers.

The crystal structure of Cs4P2O7(H2O)4 in a projection along [100]. Displacement ellipsoids and colour codes are as in Fig. 2. The inset shows the diphosphate group with atom labelling
The diphosphate anion has a staggered conformation with a P–O–P angle of 128.49(10)°. Characteristic for condensed phosphate anions [27], the two P–O bond lengths to the bridging O atom are significantly longer (1.6350(13) and 1.6458(16) Å) than the terminal P–O bond lengths (averaged values 1.518 (P1) and 1.516 (P2) Å) for the two tetrahedra of the anion. The dihedral angle between the atoms of the “backbone” of the anion (O3–P1–O4; O4–P2–O7) and the P···P distance amount to 43.23(12)° and 2.9549(8) Å, respectively. All these values are in good agreement with those of the anions of the other known hydrated alkali diphosphates, viz. Na4P2O7(H2O)10 [28] (staggered conformation; average P–O terminal bond length 1.523 Å, bridging P–O 1.612 Å, P–O–P angle 130.2°; dihedral angle within the backbone 42.0°, P···P distance 2.925 Å) and K4P2O7(H2O)3 [29] (staggered conformation; 1.503, 1.636 Å, P–O–P angle 130.3°, 25.8°, P···P distance 2.968 Å).
The coordination numbers of the four caesium cations in Cs4P2O7(H2O)4 range from eight to ten. According to their location either in the diphosphate layer or in the water-containing layer, the cations display different coordination environments. Cs1 (CN = 8) is bonded to seven O atoms from three neighbouring diphosphate groups and to one water molecule in the adjacent layer, and Cs2 is bonded to eight O atoms from three diphosphate groups and to two water molecules. On the other hand, Cs3 and Cs4 each have six water molecules and three atoms from two and three diphosphate groups, respectively, as bonding partners. The four water molecules are either bonded to four (in case of OW1, OW2, OW3) or to three (OW4) caesium cations. Although H atoms of water molecules could not be located from the current data set, O···O distances between water O atoms and phosphate O atoms in the range 2.682–2.760 Å indicate hydrogen-bonding interactions of medium strengths (Table 2) that help to consolidate the crystal packing.
CsPO3
Corbridge reported crystallographic data of CsPO3 and other alkali long-chain polyphosphates of formula (MPO3)nFootnote 2 (M = Na, K, Rb, Cs), showing that the Rb and Cs catena-polyphosphates crystallize isotypically in space group type P21/n [20]. The crystal structure of the corresponding room-temperature modification of RbPO3 was subsequently determined [29] and later re-examined twice [30, 31]. The given lattice parameters from the first study of CsPO3 at room temperature (a = 12.71, b = 4.32, c = 6.99 Å, β = 83° [20]) are in good agreement with the current low-temperature data (Table 3, with β > 90° according to convention). Next to CsPO3, TlPO3 is so far the only other known catena-polyphosphate crystallizing isotypically with the RT-form of RbPO3. However, crystallographic details of the thallium phase are restricted to lattice parameter and an indexed powder diffractogram [32].
The crystal structure of CsPO3 (Fig. 8) comprises a polyphosphate chain extending parallel to [010], with a repeating unit of two phosphate tetrahedra. The bond lengths distribution is typical for polyphosphate chains [27], with two short P—O distances (average 1.486 Å) to terminal O atoms (O2, O3) and two considerably longer P-O distances (average 1.616 Å) to bridging O atoms (O1 and O1(− x + 1/2, y − 1/2, − z + 1/2)). The Cs cations are situated in-between the chains and exhibit a coordination number of 7 with a monocapped prism as coordination polyhedron; Cs‒O distances range from 3.03 to 3.37 Å (Table 1).

The crystal structure of CsPO3 in a projection along [010]. Displacement ellipsoids and colour codes are as in Fig. 1. The inset shows the polyphosphate chains with a repetition period of 2 extending parallel [010]
The program compstru [33], available at the Bilbao Crystallographic Server [34], was employed for a quantitative structural comparison of the isotypic CsPO3 and RbPO3 structures. For that purpose, the current low-temperature structure data of CsPO3 and the room-temperature structure data of RbPO3 were used, neglecting the effect of different measurement temperatures. The comparison revealed a close structural similarity between the structures. The degree of lattice distortion is 0.0193, and the distances between the atomic positions of paired atoms are 0.0211 Å for Cs/Rb, 0.0658 Å for P1, 0.0836 Å for O1, 0.1244 Å for O2, and 0.1340 Å for O3. The arithmetic mean of all distances between paired atoms is 0.0858 Å, and the measure of similarity is 0.042.
Conclusion
The crystal structures of the four caesium phosphates Cs3(H1.5PO4)2(H2O)2, Cs3(H1.5PO4)2, Cs4P2O7(H2O)4, and CsPO3 were refined from low-temperature X-ray diffraction data at − 173 °C. Although for the two hydrogen phosphates Cs3(H1.5PO4)2(H2O)2 and Cs3(H1.5PO4)2 all hydrogen atoms could clearly be located from difference Fourier maps, an uncertainty regarding the hydrogen-bonding situation between symmetry-related hydrogen phosphate tetrahedra remains. Future neutron diffraction studies are definitely required to evidence the correctness of the structure models, here in terms of the corresponding H atom positions. Neutron diffraction data may also help to determine the H atom positions of the water molecules in hydrous Cs4P2O7(H2O)4, which was not possible on basis of the current X-ray data. However, O···O distances involving the water molecules indicate the presence of two hydrogen-bonding interactions for each of the water molecules.
Experimental
Preparation
Crystals of Cs3(H1.5PO4)2 were isolated from a batch intended to produce Cs2(HPO4). To diluted phosphoric acid (≈ 5%wt), an aqueous solution of Cs2CO3 was added in the molar ratio 1:1. The mixture was carefully evaporated until dryness and kept in an oven at 130 °C for one night. Colourless plate-like crystals were isolated from the hygroscopic product that also contained bulky crystals of Cs2(HPO4). After longer contact with ambient humidity at room temperature (about 2 days), crystals of the hydrate phase Cs3(H1.5PO4)2(H2O)2 were subsequently isolated from the original reaction product.
Crystals of Cs4P2O7(H2O)4 were isolated from a batch intended to produce anhydrous Cs4P2O7 from solid state reactions. Cs4P2O7 was described as an intermediate product during thermal treatment of Cs3(H1.5PO4)2(H2O)2 [5] or Cs2(HPO4)2(H2O)2 and was reported to be polymorphic [35]. Cs2CO3 and (NH4)2HPO4 were mixed in equimolar amounts, ground and placed in a porcelain crucible that was heated from room temperature to 950 °C within 3 h, kept at that temperature for 4 h and cooled to room temperature by turning off the furnace. Colourless plates of the tetrahydrate were harvested from the bulk product.
CsPO3 was prepared by mixing a diluted solution of phosphoric acid (≈ 5%wt) and an aqueous solution of Cs2CO3 in the molar ratio of 2:1. The mixture was subsequently warmed until dryness and heated within 2 h to 750 °C, kept at that temperature for 2 h, cooled within 10 h to 300 °C and then quickly removed from the furnace.
Structure determination
All crystals were hygroscopic and thus were embedded in perfluorinated oil for protection. Diffraction experiments on optically preselected crystals followed standard measurement procedures with corresponding software packages for data collection and data reduction [36]. All data sets were corrected for absorption effects by using the semiempirical multi-scan method [37]. The crystal structures were solved by charge flipping [38] and refined with JANA2006 [39].
All H atoms in the structures of Cs3(H1.5PO4)2 and Cs3(H1.5PO4)2(H2O)2 were located from difference Fourier maps and were refined with O‒H distance constraints of 0.86(2) Å, except for the symmetrical hydrogen bond in Cs3(H1.5PO4)2 where the H atom is located on Wyckoff position 2b (2/m). H atoms of the water molecules could not be localized reliably for Cs4P2O7(H2O)4 and therefore are not included in the final crystal structure model. The CsPO3 crystal under investigation was twinned by mirroring at (100). Reflections of the individuals were separated and processed as HKLF5 data. The refined ratio for the two twin domains was 0.7480(6):0.2520(6). Coordinates and atom numbering of CsPO3 were adapted from the isotypic room-temperature modification of RbPO3 [31].
Details of the data collections and structure refinements are gathered in Table 3. Further details of the crystal structure investigations may be obtained from The Cambridge Crystallographic Data Centre (CCDC) on quoting the depository numbers listed at the end of Table 3. The data can be obtained free of charge via http://www.ccdc.cam.ac.uk/structures.
Notes
For BVS calculation of the caesium cations, Cs–O bonds up to 3.6 Å were considered as relevant because a bond of this length still adds 0.04 valence units to the overall BVS.
The term polyphosphate is established in the literature [27] but bears some ambiguity. In a strict sense, “poly” could describe any phosphate with a large number of, yet not exclusively containing Q2 phosphate groups, where the number (here 2) designates the vertices of a PO4 tetrahedron that are shared with a neighbouring PO4 tetrahedron. The prefix “meta” conveys that there are exclusively Q2 phosphate groups in the crystal structure, which is obviously true for all (MPO3)n compounds. Hence, these compounds are also referred to as catena-metaphosphates.
References
Paschos O, Kunze J, Stimming U, Maglia F (2011) J Phys Condens Matter 23:234110
Haile SM, Chisholm CRI, Sasaki K, Boysen DA, Uda T (2007) Faraday Discuss 134:17
Kim G, Griffin JM, Blanc F, Haile SM, Grey CP (2015) J Am Chem Soc 137:3867
Nikiforov AV, Berg RW, Bjerrum NJ (2018) Ionics 24:2761
Ponomareva V, Bagryantseva I, Zakharov B, Bulina N, Lavrova G, Boldyreva E (2017) Acta Crystallogr C 73:773
Voronin VI, Berger IF, Proskurnina NV, Sheptyakov DV, Goshchitskii BN, Burmakin EI, Stroev SS, Shekhtman GS (2008) Neorg Mater 44:740
Stöger B, Weil M (2014) Acta Crystallogr C 70:7
Iwata Y, Koyano N, Shibuya I (1980) J Phys Soc Jpn 49:304
Ueso Y, Kobayashi J (1976) Phys Status Solidi A 34:475
Preisinger A, Mereiter K, Bronowska W (1994) Mater Sci Forum 166:511
Yamada K, Sagara T, Yamane Y, Ohki H, Okuda T (2004) Solid State Ion 175:557
Schuele PJ, Thomas R (1985) Jpn J Appl Phys 24:935
Efremov VA, Trunov VK, Matsichek I, Gudinitsa EN, Fakeev AA (1981) Zh Neorg Khim 26:3213
Larbot A, Durand J, Norbert A (1980) Rev Chim Mineral 17:5548
Averbuch-Pouchot MT, Durif A (1993) C R Acad Sci Ser II 316:41
Averbuch-Pouchot MT, Durif A (1989) C R Acad Sci Ser II 308:1699
Tordjman I, Masse R, Guitel JC (1977) Acta Crystallogr B 33:585
Averbuch-Pouchot MT, Durif A (1986) Acta Crystallogr C 42:131
Brühne B, Jansen M (1993) Z Anorg Allg Chem 619:1633
Corbridge DEC (1955) Acta Crystallogr 8:520
Brese NE, O’Keeffe M (1991) Acta Crystallogr B 47:192
Brown ID (2002) The chemical bond in inorganic chemistry: the bond valence model. Oxford University Press, Oxford
Troyanov SI, Snigireva EM, Kemnitz E (2000) Z Kristallogr 215:364
Schroffenegger M, Eder F, Weil M, Stöger B, Schwendtner K, Kolitsch U (2019) J Alloys Compd 820:153369
Pollitt S, Weil M (2014) Z Anorg Allg Chem 640:1622
Ruiz-Martínez A, Alvarez S (2009) Chem Eur J 15:7470
Durif A (1995) Crystal chemistry of condensed phosphates. Plenum Press, New York
McDonald WS, Cruickshank DWJ (1967) Acta Crystallogr 22:43
Corbridge DEC (1956) Acta Crystallogr 9:308
Cruickshank DWJ (1964) Acta Crystallogr 17:681
Holst C, Schmahl WW, Fuess H (1994) Z Kristallogr 209:322
El Horr N (1991) J Solid State Chem 90:386
de la Flor G, Orobengoa D, Tasci E, Perez-Mato JM, Aroyo MI (2016) J Appl Cryst 49:653
Aroyo MI, Perez-Mato JM, Capillas C, Kroumova E, Ivantchev S, Madariaga G, Kirov A, Wondratschek H (2006) Z Kristallogr 221:15
Lavrova GV, Bulina NV, Min’kov VS, Matvienko AA (2016) Russ J Inorg Chem 61:284
Apex-2 and Saint (2012) Bruker AXS Inc, Madison, Wisconsin
Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D (2015) J Appl Crystallogr 48:3
Palatinus L, Chapuis G (2007) J Appl Crystallogr 40:786
Petříček V, Dušek M, Palatinus L (2014) Z Kristallogr 229:345
Becker PJ, Coppens P (1974) Acta Crystallogr A 30:129
Acknowledgements
We thank Andreas Löffler for help during preparative work on the title compounds.
Funding
Open access funding provided by TU Wien (TUW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weil, M., Stöger, B. The caesium phosphates Cs3(H1.5PO4)2(H2O)2, Cs3(H1.5PO4)2, Cs4P2O7(H2O)4, and CsPO3. Monatsh Chem 151, 1317–1328 (2020). https://doi.org/10.1007/s00706-020-02675-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-020-02675-6