
Abstract
The structure–activity relationships of newly prepared caffeates, containing nitroxide moieties as potential antioxidants have been investigated and compared to those of known natural (caffeic acid phenethyl ester, CAPE) and unnatural (N-acetylcysteine, paramagnetic alcohols) antioxidants. The in vitro antioxidant activity was tested by 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) radical scavenging, while assays of cell protection against reactive oxygen species were carried out in the presence of 2′,7′-dichlorofluorescein diacetate and H2O2. Paramagnetic esters without phenol motifs exhibited the lowest antioxidant activity, followed by paramagnetic alcohols with moderate activity. Among the compounds investigated, paramagnetic phenolic compounds were the best antioxidants. As the new paramagnetic CAPE analogs were less cytotoxic than CAPE at 10 µM in NIH3T3 fibroblast cells but had similar antioxidant activity, they can be considered promising antioxidants.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Caffeic acid phenethyl ester [2-phenylethyl (2E)-3-(3,4-dihydroxyphenyl)acrylate; CAPE, 1] is a phenolic compound isolated from the propolis of honeybee hives [1]. Although CAPE is hydrolyzed to caffeic acid in vivo, it has a better pharmacokinetic profile than that of caffeic acid [2]. CAPE possesses many biological activities, such as antioxidant [3,4,5], anti-inflammatory [6, 7], immunomodulatory [8], antiviral, and neuroprotective activities (Fig. 1) [9, 10].

Covalent structure of caffeic acid phenethyl ester (CAPE)
The phenylpropanoid scaffold of caffeic acid (3,4-dihydroxycinnamic acid) is commonly used as a template to develop caffeic acid derivatives and new chemical entities possessing antioxidant properties. Wang et al. investigated the relationship between catechol ring modifications (fluorination, altered numbers of hydroxyl groups) and the cytoprotective, cytotoxic, and antioxidant properties of CAPE [11, 12]. Their results showed that at least one hydroxyl group is important for cytoprotection and that the catechol ring is required for antioxidant activity. CAPE inhibits the HIV-1 integrase enzyme, and this activity was enhanced with the insertion of a third hydroxyl group into the catechol ring [13]. The modification of the phenethyl side also may be beneficial, as caffeic acid 4-aminophenethyl ester showed better cytoprotection and 4-nitrophenethyl ester showed better cardioprotection, while the octyl ester showed better antileukemic activity than CAPE itself [14,15,16].
The unpaired electron of nitroxide stable free radicals allows them to take part in one-electron reactions such as oxidations and reductions, which makes nitroxides potent unnatural antioxidants [17]. Nitroxides may also function as intra- and extracellular superoxide dismutase (SOD) mimics or nonthiol radioprotectants [18,19,20]. Studies indicate that as antioxidants, nitroxides can exert cytoprotective action against oxidative stress induced by cytotoxic drugs or pathological processes such as ischemia–reperfusion and inflammation [21,22,23]. However, some piperidine-type nitroxides also exert pro-oxidative and cytotoxic effects, in particular on cancer cells [24, 25].
Several studies indicated that biomolecules (resveratrol, curcumin) modified with nitroxides had a beneficial influence on their activity as supplemented by ‘in statu nascendi acting’ antioxidants and radical scavengers [26,27,28,29,30]. Herein, we report the synthesis, antioxidant and cytoprotective activity of new CAPE analogs compared with those of the parent compound (CAPE) and regular antioxidant standards, such as N-acetylcysteine and Trolox. The antioxidant and cell protection activity of paramagnetic building block alcohols 3-(hydroxymethyl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-oxyl (2), 4-(hydroxymethyl)-2,2,6,6-tetramethyl-5,6-dihydropyridin-1(2H)-oxyl (3) (Fig. 2) were also evaluated [31, 32]. To obtain new paramagnetic analogs of CAPE, three strategies were followed: (1) replacement of the catechol ring with nitroxides, (2) replacement of the phenethyl group with nitroxide rings, and (3) incorporation the pyrroline radical in the caffeic acid scaffold.

Structure of paramagnetic alcohols to be investigated
Results and discussion
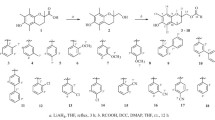
The Horner–Wadsworth–Emmons reaction of paramagnetic aldehydes 1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carbaldehyde (4) [31] with triethyl phosphonoacetate and 1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridine-4-carbaldehyde (5) [32] with trimethyl phosphonoacetate led to the formation of paramagnetic α,β-unsaturated esters ethyl (E)-3-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)acrylate (6) [33] and methyl (E)-3-(1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)acrylate (7), respectively. Their hydrolysis was conducted at room temperature for 2 h with a 10% aq. NaOH/methanol solution (2.0 equiv. NaOH) to yield, after acidification with aq. H2SO4, carboxylic acids (E)-3-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)acrylic acid (8) [31] and (E)-3-(1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)acrylic acid (9), respectively. Then, the paramagnetic carboxylic acids were treated with 2-phenethyl bromide in dry acetonitrile in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to obtain compounds phenethyl (E)-3-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)acrylate (10) and phenethyl (E)-3-(1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)acrylate (11), in which the catechol ring of CAPE is replaced with dihydropyrrole- or tetrahydropyridine-type nitroxides (Scheme 1).

For the replacement of the phenethyl moiety of CAPE, the first step was to alkylate (E)-3-(3,4-diacetoxyphenyl)acrylic acid (12) [34] with paramagnetic allylic bromides 3-(bromomethyl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxy (13) [35] and 4-(bromomethyl)-2,2,6,6-tetramethyl-5,6-dihydropyridin-1-yloxy (14) [36]. The alkylation was conducted in CH3CN in the presence of DBU base at ambient temperature to give compounds (1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-yl)methyl (E)-3-(3,4-diacetoxyphenyl)acrylate (15) and (1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl (E)-3-(3,4-diacetoxyphenyl)acrylate (16) in 60–71% yield. To obtain paramagnetic caffeic acid esters (1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-yl)methyl (E)-3-(3,4-dihydroxyphenyl)acrylate (17) and (1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl (E)-3-(3,4-dihydroxyphenyl)acrylate (18), the acetyl protecting groups were removed by the addition of three equivalents K2CO3 in CH2Cl2/MeOH mixture (Scheme 2).

To keep both catechol and phenethyl groups, the carbon–carbon double bond of CAPE seemed to be an accessible point for the synthesis of a new paramagnetic derivative. 4-Bromo-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carboxylic acid (19) [37] was alkylated with 2-phenethyl bromide to yield phenethyl 4-bromo-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-carboxylate (20). The Suzuki reaction of 20 and 2-[3,4-bis(methoxymethoxy)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (21) [38] in the presence of Pd(PPh3)4 and Na2CO3 under a N2 atmosphere afforded phenethyl 4-[3,4-bis(methoxymethoxy)phenyl]-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carboxylate (22) in moderate yield. The methoxymethyl protecting groups were removed by refluxing in MeOH containing a catalytic amount of aq. HCl, resulting in nitroxide phenethyl 4-(3,4-dihydroxyphenyl)-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carboxylate (23) (Scheme 3).

The new nitroxides were tested in a Trolox-equivalent antioxidant capacity assay (TEAC), which monitors electron and proton donor activity [39, 40]. The method is based on the reduction of green-colored 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) radical (ABTS·+), which is detected at 734 nm.
Our results suggest that the catechol functionality is necessary for good antioxidant activity, as the TEAC values of paramagnetic alcohols 2 and 3 nonphenolic esters 7, 10, and 11; and acid 9 were below 0.2, while the caffeic acid derivatives CAPE (1), 17, 18, and 23 showed good antioxidant activity. Compounds 17 and 18 have even greater proton- and electron-donating properties than Trolox and CAPE. Nevertheless, in the case of compound 23, the vicinity of nitroxide and catechol had adverse effect on antioxidant activity (Fig. 3), which can be the consequence of an intramolecular redox reaction between the nitroxide and catechol rings [41].

Trolox equivalent antioxidant capacity (TEAC) presents the antioxidant activity of nitroxides measured by ABTS radical scavenging assay. Data represent the mean ± SD (n = 6)
Cytotoxicity of CAPEs was analyzed by the WST assay. As shown in Fig. 4, 10 µM CAPE (1) decreased cellular viability (0.63 ± 0.09, relative to DMSO), and the reference compound N-acetylcysteine (NAC) increased it (1.36 ± 0.24). We did not observe any statistical significance in the difference in cellular viability between the CAPE analogs and DMSO; thus, these new compounds have less cytotoxic activity than CAPE. Compounds 1, 17, 18, and 23 exerted some toxicity toward fibroblasts, while the paramagnetic alcohols and nonphenolic esters were not toxic. This result suggests the responsibility of the catechol ring for cytotoxicity (Fig. 4), but this is still tolerable.

Cytotoxicity of CAPEs were analyzed by WST assay. Cellular viability of NIH3T3 cells treated with nitroxides, CAPE or NAC (10 μM) for 24 h. Data represent the mean ± SD (n = 6). *Statistically significant (p < 0.05) when compared to control
The intracellular oxidative stress within NIH3T3 cells was evaluated by 2′,7′-dichlorofluorescein diacetate (DCFDA) [42, 43], which is taken up by the cells, wherein nonspecific esterases cleave the acyl groups, forming 2′,7′-dichlorodihydrofluorescein (H2DCF), which can be oxidized to fluorescent DCF by reactive oxygen species (ROS). Therefore, the limited formation of DCF (and fluorescence intensity) indicates the enhanced ROS scavenging activity of the investigated compound. All of the studied compounds decreased the ROS formation in cells: CAPE (1), 17, 18, and NAC significantly decreased intracellular oxidative stress (Fig. 5). This activity cannot be attributed to the presence of nitroxide functionality, as compounds 2, 3, and 23 were not active, but retaining the original caffeic acid ester scaffold was advantageous, as in the case of compounds 1, 17, and 18.

Intracellular oxidative stress was evaluated by the fluorescence intensity of 2′,7′-dichlorofluorescein. Cells were treated with 10 µM each compound for 24 h. After treatment, cells were stained with 20 µM DCFDA for 30 min at 37 °C. Data represent the mean ± SD (n = 6). *Statistically significant (p < 0.05) when compared to control
Reactive oxygen species are involved in the pathogenesis of many diseases. The hydroxyl radical is the most dangerous ROS and can be generated from H2O2 in the presence of transition metal ions including Fe2+ and Cu+ in certain processes. To confirm the antioxidant properties of new caffeic acid phenethyl ester analogs, the cytoprotective effect of the paramagnetic CAPEs against hydrogen peroxide (H2O2) was tested. H2O2 treatment decreased cellular viability (0.41 ± 0.08), but CAPE (1), 17, and 23, containing caffeic acid ester scaffold, prevented H2O2-induced cytotoxicity. It is interesting to note that compounds 7, 9, 10, and 11 did not exhibit antioxidant protection in this assay, while alcohols 2, 3 and compound 18 produced a moderate protective effect (Fig. 6).

To determine the cytoprotective activity of CAPEs toward H2O2 were analyzed using WST assay. Cells were treated with H2O2 (500 µM) with CAPEs (10 µM), respectively, for 24 h. Data represent the mean ± SD (n = 6). *Statistically significant (p < 0.05) when compared to control
Conclusion
Three structural fragments of caffeic acid phenethyl ester were modified with nitroxide free radicals to study their antioxidant structure–activity relationships. The main modifications were the replacement of the catechol ring or phenethyl group with 5- and 6-membered nitroxides and the incorporation of pyrrolidine type of nitroxide into the CC double bond of the acrylate. The toxicity and antioxidant properties of the new compounds were compared to those of CAPE (1) and regular antioxidant standards.
Regarding the ABTS assay, paramagnetic nonphenolic esters 7, 10, and 11 and alcohols 2 and 3 have poor ABTS·+ scavenging activity, which is not surprising because esters can be electron donors but not proton donors. As expected, compounds 1, 17, 18, and 23 were found to be good proton and electron donor antioxidants.
The results of the cytotoxicity assays suggest that the catechol ring motif increases cytotoxicity, but not significantly. The most toxic compounds were CAPE (1) and compound 23, but when nitroxides were the esterifying groups, as in the case of compounds 17 and 18, the toxicity of the resulting compound was lower than that of 1 and 23. In DCF assays, compounds 1, 17, and 18 were the most active in intracellular ROS scavenging and had the same or slightly greater antioxidant effects than NAC. In cell viability assays performed under H2O2-induced stress, compounds 1, 17, and 23 were the most protective molecules, as all showed better activity than the standard (NAC). However, the results of the assays of nitroxide derivatives were contradictory, as compounds 17 and 18 with nitroxide and catechol structural units were the best proton and electron donor antioxidants and the most protective against H2O2-induced cytotoxicity.
These findings support our hybrid drug design strategy and are in good accordance with our previous findings. The caffeic acid moiety should remain intact, and proper esterification, in our case with nitroxides, decreases its cytotoxicity while simultaneously improving its protective activity against H2O2−-induced oxidative stress. Our results can be a good starting point in the search for new caffeic acid-based lead compounds, which is in progress in our laboratories.
Experimental
Elemental analyses were performed on a Fisons EA 1110 CHNS elemental analyzer, and the results were found to be in good agreement (± 0.3%) with the calculated values. Melting points were determined with a Boetius micro-melting point apparatus. Mass spectra were recorded on a Thermoquest Automass Multi in EI mode (70 eV). 1H NMR spectra were recorded with Bruker Avance 3 Ascend 500 at 500 MHz and 13C NMR spectra at 125 MHz in CDCl3 or DMSO-d6 at 298 K in the presence of five equivalents of hydrazobenzene (DPPH/radical). The IR spectra were taken with a Bruker Alpha FT-IR instrument with ATR support on a ZnSe plate. Spectroscopic measurements were performed on Specord 40 UV/VIS Spectrophotometer at 732 nm. Flash column chromatography was performed on Merck Kieselgel 60 (0.040–0.063 mm). Qualitative TLC was carried out on commercially available plates (20 × 20 × 0.02 cm) coated with Merck Kieselgel GF254. Compounds 1 [34], 2 [31], 3 [32], 4 [31], 5 [32], 12 [33], 13 [34], 14 [35], 19 [36], and 21 [37] were synthesized according to published procedures. All other reagents were purchased from Aldrich or Molar Chemicals.
(E)-Methyl 3-(1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)acrylate (7, C13H20NO3)
To a stirred suspension of 0.3 g NaH (12.5 mmol) in 10 cm3 THF 910 mg trimethyl phosphonoacetate (5.0 mmol) was added at 0 °C under a N2 atmosphere. After stirring for 10 min, 910 mg aldehyde 5 (5.0 mmol) was added, and the mixture was stirred and refluxed for another 30 min. After cooling to room temperature, 20 cm3 brine and 20 cm3 ether were added, the organic phase was separated, the aqueous phase was washed with 2 × 20 cm3 ether, and then the combined organic phase was dried (MgSO4), filtered, evaporated, and the residue was purified by flash column chromatography to give 655 mg (55%) 7 as a pale orange solid. M.p.: 66–68 °C; Rf = 0.55 (hexane–EtOAc, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.38 (d, J = 15.8 Hz, 1H), 5.93 (s, 1H), 5.86 (d, J = 15.7 Hz, 1H), 3.82 (s, 3H), 2.26 (s, 2H), 1.35 (s, 6H), 1.26 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 167.7, 146.8, 144.3, 128.5, 116.1, 60.1, 57.2, 51.6, 38.9, 25.9, 25.1 ppm; IR (neat): \(\bar{\nu }\) = 1712, 1642, 1624 cm−1; MS (70 eV): m/z (%) = 238 (M+, 93), 208 (28), 193 (30), 149 (100).
(E)-3-(1-Oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)acrylic acid (9, C12H18NO3)
To a solution of 1.19 g 7 (5.0 mmol) in 30 cm3 MeOH, 4 cm3 aq. 10% NaOH (10.0 mmol) was added, and the mixture stirred for 2 h at ambient temperature. The mixture was diluted with 10 cm3 water, the methanol was evaporated off, and the residue pH was adjusted to 2 at 0 °C by adding aq. 5% H2SO4 solution. The acid formed was extracted with 2 × 2 cm3 CHCl3, and the combined organic phase was dried (MgSO4), filtered, and evaporated. The residue was purified by flash column chromatography (hexane–EtOAc, 2:1) to give 870 mg (78%) compound 9 as a yellow solid. M.p.: 156–158 °C; Rf = 0.25 (CHCl3–Et2O, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.25 (d, J = 16 Hz, 1H), 6.01 (d, J = 15.7 Hz, 1H), 5.84 (s, 1H), 2.59 (s, 2H), 1.49 (s, 6H), 1.42 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 173.6, 142.1, 136.5, 125.5, 123.6, 64.4, 62.3 36.5, 23.9 (brs, 4C) ppm; IR (neat): \(\bar{\nu }\) = 3250–2500, 1671, 1613 cm−1; MS (70 eV): m/z (%) = 224 (M+, 76), 194 (32), 179 (72), 149 (100).
General procedure for the synthesis of phenethyl esters 10, 11, 20
To the solution of 212 mg 8 or 224 mg 9 or 263 mg 19 (1.0 mmol) in 5 cm3 CH3CN, 167 mg DBU (1.1 mmol), and 277 mg (2-bromoethyl)benzene (1.1 mmol) were added, and the mixture was allowed to react for 24 h at room temperature. Then, the solvent was evaporated, and the residue was purified by flash column chromatography (hexane–EtOAc, 4:1) to give the title compounds in 65–70% yield.
Phenethyl (E)-3-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)acrylate (10, C19H24NO3)
Dark yellow solid; yield 233 mg (70%); m.p.: 61–63 °C; Rf = 0.59 (hexane–EtOAc, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.37 (t, J = 6.9 Hz, 2H), 7.26–7.22 (m, 3H), 6.15 (d, J = 16.3 Hz, 1H), 5.99 (s, 1H), 4.46 (t, J = 7.1 Hz, 2H), 3.05 (t, J = 7.1 Hz, 2H), 1.41 (s, 6H), 1.32 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 166.9, 141.2, 140.1, 138.6, 137.9, 129.4, 128.6, 126.7, 119.9, 69.7, 67.7, 65.1, 35.3, 25.5, 24.9 ppm; IR (neat): \(\bar{\nu }\) = 1698, 1603, 1455 cm−1; MS (70 eV): m/z (%) = 314 (M+, 6), 299 (5), 284 (18), 105 (100), 91 (44).
Phenethyl (E)-3-(1-oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)acrylate (11, C20H26NO3)
Orange solid; yield 215 mg (65%); m.p.: 60–61 °C; Rf = 0.60 (hexane–EtOAc, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.37 (d, J = 6.6 Hz, 2H), 7.27 (t, J = 7.3 Hz, 3H), 5.91 (s, 1H), 5.83 (d, J = 15.8 Hz, 1H), 4.44 (t, J = 7.1 Hz, 2H), 3.04 (t, J = 7.1 Hz, 2H), 2.24 (s, 2H), 1.34 (s, 6H), 1.25 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 167.2, 146.8, 144.3, 137.9, 129.4, 128.55, 128.53, 126.6, 116.3, 64.9, 60.1, 57.2, 38.9, 35.3, 26.1, 25.0 ppm; IR (neat): \(\bar{\nu }\) = 1705, 1636, 1617 cm−1; MS (70 eV): m/z (%) = 328 (M+, 2), 179 (5), 149 (31), 105 (100).
Phenethyl 4-bromo-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-carboxylate (20, C17H21BrNO3)
Yellow solid; yield 256 mg (70%); m.p.: 77–79 °C; Rf = 0.71 (hexane–EtOAc, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.32 (t, J = 7.4 Hz, 2H), 4.46 (t, J = 7.1 Hz, 2H), 3.04 (t, J = 7.1 Hz, 2H), 1.34 (s, 6H), 1.32 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 163.1, 137.6, 136.9, 134.3, 129.4, 128.9, 128.6, 126.7, 72.0, 70.6, 65.3, 35.0, 24.5, 24.4 ppm; IR (neat): \(\bar{\nu }\) = 1704, 1624, 1592 cm−1; MS (70 eV): m/z (%) = 368 (M+, 15), 262 (5), 232 (6), 105 (100), 91 (83).
General procedure for the synthesis of protected, paramagnetic caffeic acids 15, 16
Caffeic acid diacetate 12 (2.64 g, 10.0 mmol), 1.67 g DBU (11.0 mmol), and 2.33 g 13 or 2.47 g 14 (10.0 mmol) were dissolved in 15 cm3 CH3CN and the mixture was allowed to react for 24 h at ambient temperature. The solvent was then evaporated, and the residue was purified by flash column chromatography (hexane–EtOAc, 4:1) to give the title compounds in 60–71% yield.
(1-Oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-yl)methyl (E)-3-(3,4-diacetoxyphenyl)acrylate (15, C22H26NO7)
Pale yellow solid; yield 2.95 g (71%); m.p.: 132–135 °C; Rf = 0.59 (CHCl3-Et2O, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.73 (d, J = 16 Hz, 1H), 7.48–7.45 (m, 2H), 6.48 (d, J = 16 Hz, 1H), 5.69 (s, 1H), 4.81 (s, 2H), 2.37 (2 s, 6H), 1.37 (s, 6H), 1.33 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 168.0, 167.9, 166.2, 143.7, 143.3, 142.6, 139.3, 133.2, 132.4, 126.5, 123.9, 122.8, 119.9, 69.9, 67.8, 60.8, 25.6, 24.6, 20.6 ppm; IR (neat): \(\bar{\nu }\) = 1766, 1713, 1634 cm−1; MS (70 eV): m/z (%) = 416 (M+, 10), 317 (2), 205 (16), 105 (75), 43 (100).
(1-Oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl (E)-3-(3,4-diacetoxyphenyl)acrylate (16, C23H28NO7)
Orange solid; yield 2.58 g (60%); m.p.: 57–59 °C; Rf = 0.64 (CHCl3–Et2O, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.72 (d, J = 15.9 Hz, 1H), 7.46–7.43 (m, 2H), 6.48 (d, J = 15.9 Hz, 1H), 5.61 (s, 1H), 4.65 (s, 2H), 2.34 (2 s, 6H), 2.18 (s, 2H), 1.34 (s, 6H), 1.28 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 168.1, 167.9, 166.4, 143.6, 143.1, 142.5, 133.5, 133.3, 126.6, 126.5, 123.9, 122.8, 119.2, 67.7, 59.5, 57.4, 40.8, 26.17, 25.12 ppm; 20.66, 20.63 IR (neat): \(\bar{\nu }\) = 1765, 1709, 1503 cm−1; MS (70 eV): m/z (%) = 430 (M+, 2), 247 (9), 205 (36), 163 (38), 121 (79), 43 (100).
General procedure for the synthesis of paramagnetic caffeic acids 17, 18
15 (2.08 g) or 16 (2.15 g) (5.0 mmol) was dissolved in 20 cm3 MeOH–CH2Cl2 (1:1), 2.07 g K2CO3 (15.0 mmol) was added, and the mixture was stirred at 20 °C for 1 h. The solvent was evaporated, and the residue was partitioned between 10 cm3 5% H2SO4 and 20 cm3 EtOAc. After separation, the water phase was extracted with additional 2 × 20 cm3 EtOAc. The combined organic phases were dried over MgSO4, filtered, and evaporated, and the residue was purified by column chromatography (hexane–EtOAc) to obtain paramagnetic caffeic acids in 67–72% yield.
(1-Oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-yl)methyl (E)-3-(3,4-dihydroxyphenyl)acrylate (17, C18H22NO5)
Yellow solid; yield 1.19 g (72%); m.p.: 123–125 °C; Rf = 0.36 (CHCl3-Et2O, 2:1); 1H NMR (500 MHz, DMSO-d6 + (PhNH)2): δ = 7.03 (d, J = 5.1 Hz, 1H), 6.81 (d, J = 6.2 Hz, 1H), 6.6 (s, 1H), 6.34 (d, J = 15.3 Hz, 1H), 5.60 (s, 1H), 5.83 (d, J = 15.8 Hz, 1H), 4.69 (s, J = 7 Hz, 2H), 1.16, 1.12 (2 s, 12H) ppm; 13C NMR (125 MHz, DMSO-d6 + (PhNH)2): δ = 166.6, 150.4, 146.1, 145.9, 140.3, 125.9, 121.9, 118.2, 116.3, 115.4, 114.2, 68.9, 66.9, 60.4, 26.1, 25.0 ppm; IR (neat): \(\bar{\nu }\) = 3480–2700, 1717, 1603 cm−1; MS (70 eV): m/z (%) = 332 (M+, 3), 317 (1), 182 (72), 107 (100).
(1-Oxyl-2,2,6,6-tetramethyl-1,2,3,6-tetrahydropyridin-4-yl)methyl (E)-3-(3,4-dihydroxyphenyl)acrylate (18, C19H24NO5)
Orange-brownish solid; yield 1.16 g (67%); m.p.: 74–76 °C; Rf = 0.37 (CHCl3-Et2O, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.12 (s, 1H), 7.1 (d, J = 7.5 Hz, 1H), 6.28 (d, J = 15.8 Hz, 1H), 5.63 (s, 1H), 4.61 (s, 2H), 1.73 (s, 2H), 1.40 (s, 6H), 1.32 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 167.4, 147.3, 145.8, 144.7, 131.5, 127.3, 127.1, 122.5, 115.6, 114.7, 114.4, 67.1, 61.5, 59.5, 39.7, 25.6, 24.5 ppm; IR (neat): \(\bar{\nu }\) = 3500–2800, 1694, 1597 cm−1; MS (70 eV): m/z (%) = 346 (M+, 4), 307 (5), 234 (26), 163 (100).
Phenethyl 4-[3,4-bis(methoxymethoxy)phenyl]-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carboxylate (22, C27H34NO7)
To a deoxygenated solution of 545 mg 20 (1.5 mmol) in 20 cm3 dioxane/water (4:1), 75 mg Pd(PPh3)4 (0.067 mmol), 490 mg pinacolate 21 (1.5 mmol), and 5 cm3 10% Na2CO3 solution were added under N2 gas, and then the reaction mixture was stirred and refluxed for 3 h. After consumption of the starting materials (followed by TLC), dioxane was removed by vacuum evaporation, 10 cm3 H2O was added, and the aqueous phase was extracted with 2 × 20 cm3 CHCl3. The combined organic phase was dried (MgSO4), filtered, and evaporated. The residue was purified by flash column chromatography (hexane–EtOAc, 2:1), to offer 280 mg (58%) 22 as a brown oil. Rf = 0.38 (hexane–EtOAc 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.33 (t, J = 7.5 Hz, 2H), 7.21 (d, J = 8.3 Hz, 1H), 7.14 (d, J = 7.2 Hz, 2H), 7.07 (s, 1H), 6.82 (d, J = 6.4 Hz, 1H), 5.30 (2 s, 4H), 4.24 (t, J = 7.2 Hz, 2H), 3.58 (d, J = 3.3 Hz, 6H), 2.71 (t, J = 7.2 Hz, 2H), 1.54 (s, 6H), 1.35 (s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 164.7, 154.4, 147.1, 146.6, 137.8, 133.3, 129.9, 128.8, 128.5, 126.5, 121.2, 117.4, 116.1, 95.8, 95.5, 71.4, 69.2, 64.6, 56.28, 34.7, 24.8, 24.3 ppm; IR (neat): \(\bar{\nu }\) = 1707, 1602, 1506 cm−1; MS (70 eV): m/z (%) = 484 (M+, 6), 470 (1), 274 (30), 105 (100).
Phenethyl 4-(3,4-dihydroxyphenyl)-1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole-3-carboxylate (23, C23H26NO5)
484 mg 22 (1.0 mmol) was dissolved in 10 cm3 MeOH, then concentrated 0.1 cm3 aq. HCl was added, and the mixture was refluxed for 10 min. After removal of the solvent, 10 cm3 water was added, and the residue was neutralized with K2CO3. EtOAc (20 cm3) was added, the phases were separated, and the aqueous phase was extracted with 2 × 20 cm3 EtOAc. The combined organic phase was dried (MgSO4), filtered, evaporated, and purified by flash chromatography (hexane–EtOAc 2:1) to give 310 mg (78%) title compound 23 as a brown oil. Rf = 0.57 (CHCl3-Et2O, 2:1); 1H NMR (500 MHz, CDCl3 + (PhNH)2): δ = 7.09 (d, J = 7.3 Hz, 2H), 6.69 (s, 1H), 6.61 (s, 1H), 4.21 (t, J = 6.8 Hz, 2H), 2.69 (t, J = 6.7 Hz, 2H), 1.51 (s, 6H), 1.31 (2 s, 6H) ppm; 13C NMR (125 MHz, CDCl3 + (PhNH)2): δ = 164.3, 154.7, 144.2, 143.2, 137.6, 132.7, 128.8, 128.5, 127.3, 126.5, 119.5, 115.7, 114.9, 71.7, 69.7, 64.8, 34.7, 24.8, 23.8 ppm; IR (neat): \(\bar{\nu }\) = 3600–2850, 1703, 1601, 1515 cm−1; MS (70 eV): m/z (%) = 396 (M+, 49), 381 (5), 366 (6), 105 (100).
ABTS scavenging assay
The measurements were taken on a Specord 40 instrument. ABTS was dissolved in PBS buffer (0.136 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, 0.00176 M KH2PO4) to a 7-mM concentration. ABTS radical cation (ABTS•+) was produced by reacting ABTS stock solution with potassium persulfate at a final concentration of 2.45 mM and allowing the mixture to stand in the dark at room temperature for 16 h before use. For the study of compounds, the ABTS·+ solution was diluted with ethanol to an absorbance of 0.70 (± 0.02) at 734 nm and equilibrated at 37 °C. Stock solutions of new compounds and Trolox in DMSO were added to the diluted ABTS·+ solution, in final concentrations of 12.5, 10, 7.5, 2.5 µM. After addition, the mixtures were incubated for 6 min at 37 °C before measuring their absorbance at 734 nm. All determinations were carried out three times. The percentage inhibition of absorbance at 734 nm is calculated with the usual formula: (A0 − Aantioxidant)/A0, where A0 is the absorbance of the diluted ABTS·+ solution. The concentration–response curves of new compounds were compared with the curve of Trolox.
Cell viability assays
Murine embryonic fibroblast NIH3T3 cells were maintained in DMEM (Cat no. 043-30085, Wako Pure Chemical, Osaka, Japan) supplemented with 10% newborn calf serum (NBCS) at 37 °C, 5% CO2.
To determine the toxicity of the compounds, a WST-1 viability assay was employed as described previously. NIH3T3 cells (2000 per well) were seeded into all wells of 96-well plates. After 24-h incubation, the medium was replaced with CAPEs-containing medium (10 µM) for 24 h. Then, the cells were treated with WST-1 solution [3.6 µg/mm3 WST-1, 70 ng/mm3 1-methoxyphenazine methosulfate in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesufonic acid–KOH (pH 7.4); Dojindo, Kumamoto, Japan]. The cells were incubated for 1 h at 37 °C, and the absorbance of each well was recorded at 440 nm using a Multiskan FC microplate reader (Thermo Fisher Scientific).
To determine the cytoprotective activity of CAPEs against H2O2, NIH3T3 cells were treated with H2O2 (500 µM) and CAPEs (10 µM). After 24 h of incubation, cellular viability was analyzed as described above [44].
Intracellular ROS detection
Intracellular oxidative stress was evaluated by 2′,7′-dichlorofluorescein diacetate (DCFDA; Cat NO. 6883, Sigma-Aldrich) staining as previously reported. NIH3T3 cells were treated with each compound at 10 µM for 24 h. After treatment, the cells were stained with 20 µM DCFDA for 30 min at 37 °C. Then, the cells were trypsinized, and their fluorescence intensity was analyzed using an EC800 Analyzer (Sony Corporation, Tokyo, Japan).
Statistical analyses were done by Dunnett’s test.
References
Grunberger D, Banerjee R, Eisinger K, Oltz EM, Efros L, Caldwell M, Estevez V, Nakanishi K (1988) Experientia 44:230
Wang X, Pang J, Maffucci JA, Pade DS, Newman RA, Kerwin SM, Bowman PD, Stavchansky S (2009) Biopharm Drug Dispos 30:221
Wu WM, Lu L, Long Y, Wang T, Liu L, Chen Q, Wang R (2007) Food Chem 105:107
Murtaza G, Karim S, Akram MR, Khan SA, Azhar S, Mumatz A, Bin Asad MH (2014) Biomed Res Int 145342:1
Russo A, Longo R, Vanella A (2002) Fitoterapia 73:21
Juman S, Yasui J, Ikeda K, Ueda A, Sakanaka M, Negishi H, Miki T (2012) Biol Pharm Bull 35:1941
Toyoda T, Tsukamoto T, Takasu S, Shi L, Hirano N, Ban H, Kumagai T, Tatematsu M (2009) Int J Cancer 73:21
Park JH, Lee JK, Kim HS, Chung ST, Eom JH, Kim KA, Chung SJ, Paik SY, Oh HY (2004) Int Immunopharmacol 4:429
Uzar E, Sahin O, Koyuncuoglu HR, Uz E, Bas O, Kilbas S, Yilmaz HR, Yurekli VA, Kucuker H, Songur A (2006) Toxicology 218:125
Kumar M, Kaur D, Bansal N (2017) Pharmacogn Mag 13:10
Wang X, Stavchansky S, Bowman PD, Kerwin SM (2006) Bioorg Med Chem 14:4879
Wang X, Stavchansky S, Kerwin SM, Bowman PD (2010) Eur J Pharm 635:16
Burke TR, Fesen MR, Mazumder A, Wang J, Carothers AM, Grunberger D, Driscoll J, Pommier Y (1995) J Med Chem 38:4171
Shi H, Xie D, Yang R, Cheng Y (2014) J Agric Food Chem 62:5046
Du Q, Hao C, Gou J, Li X, Zou K, He X, Li Z (2016) Exp Ther Med 11:1433
Etzenhouser B, Hansch C, Kapur S, Selassie CD (2001) Bioorg Med Chem 9:199
Krishna MC, DeGraff W, Hankovszky HO, Sár PC, Kálai T, Jeko J, Russo A, Mitchell JB, Hideg K (1998) J Med Chem 41:3477
Soule BP, Hyodo F, Matsumoto KI, Simone NL, Krishna M, Mitchell JB (2007) Antioxid Redox Signal 9:1731
Likhtenshtein GI, Yamauchi J, Nakatsuji S, Smirnov AI, Tamura R (2008) Nitroxides, applications in chemistry, biomedicine, and materials science. Wiley-VCH, Weinheim
Mitchell JB, Krishna A, Samuni P, Kuppusamy P, Hahn SA, Russo A (2000) In: Rhodes CJ (ed) Toxicology of the human environment. Taylor & Francis, New York, p 113
Monti E, Cova D, Guido E, Morelli R, Oliva C (1996) Free Radic Biol Med 21:463
Howard BJ, Yatin S, Hensley K, Allen KL, Kelly JP, Carney J, Butterfield DA (1996) J Neurochem 67:2045
Deres P, Halmosi R, Tóth A, Kovács K, Pálfi A, Habon T, Czopf L, Kálai T, Hideg K, Sümegi B, Tóth K (2005) J Cardiovasc Pharmacol 45:36
Suy S, Mitchell JB, Samuni A, Mueller S, Kasid U (2005) Cancer 103:1302
Suy S, Mitchell JB, Ehleiter D, Haimovitz-Friedman A, Kasid U (1998) J Biol Chem 273:17871
Kálai T, Borza E, Antus CS, Radnai B, Gulyás-Fekete G, Fehér A, Sümegi B, Hideg K (2011) Bioorg Med Chem 19:7311
Dayton A, Selvendiran A, Meduru S, Khan M, Kuppusamy ML, Naidu S, Kálai T, Hideg K, Kuppusamy P (2011) J Pharm Exp Ther 2:350
Kálai T, Kuppusamy ML, Balog M, Selvendiran K, Rivera BK, Kuppusamy P, Hideg K (2011) J Med Chem 54:5414
Ravi Y, Selvendiran K, Naidu S, Meduru S, Citro LA, Bognár B, Khan M, Kálai T, Hideg K, Kuppusamy P, Sai-Sudhakar CB (2013) Hypertension 61:593
Bognár B, Kuppusamy ML, Madan E, Kálai T, Balog M, Jekő J, Kuppusamy P, Hideg K (2017) Med Chem 13:761
Hideg K, Hankovszky HO, Lex L, Kulcsar GY (1980) Synthesis (11):911
Csekő J, Hankovszky HO, Hideg K (1985) Can J Chem 63:940
Hankovszky HO, Hideg K, Lex L, Kulcsár GY, Halász AP (1982) Can J Chem 60:1432
Touaibia M, Guay MJ (2011) Chem Edu 88:473
Hankovszky HO, Hideg K, Lex L (1980) Synthesis 12:914
Hideg K, Csekő J, Hankovszky HO, Sohár P (1986) Can J Chem 64:1482
Chudinov AV, Rozantsev EG, Rosinov BV (1983) Izv Acad Nauk Ser Khim (2):409
Steinke N, Jahr M, Lehmann M, Baro A, Frey W, Tussetschlaeger S, Sauer S, Laschat S (2009) J Mat Chem 19:645
Re R, Pellegrini N, Prottegente A, Pannala A, Yang M, Rice-Evans A (1999) Free Radic Biol Med 26:1231
Pérez-Jiménez J, Saura-Calixto F (2008) Int J Food Sci Tech 43:185
Fuchs J, Groth N, Herrling T, Zimmer G (1997) Free Radic Biol Med 22:967
Heston AS, Brandt R (1965) Anal Biochem 11:1
Lebel CP, Ischiropoulos H, Bondy SC (1992) Chem Res Toxicol 5:227
Sakai Y, Yamamory T, Yasui H, Inanami O (2015) Biochem Biophys Res Commun 461:35
Acknowledgements
Open access funding provided by University of Pécs (PTE). We are grateful to Gergely Gulyás-Fekete (Department of Biochemistry and Medical Chemistry) for NMR measurements. The project was supported by Hungarian National, Research, Development and Innovation Office (OTKA 124331) and Grant GINOP (GINOP-2.3.2-15-2016-00049).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nagane, M., Yamashita, T., Vörös, P. et al. Synthesis and evaluation of paramagnetic caffeic acid phenethyl ester (CAPE) analogs. Monatsh Chem 150, 1513–1522 (2019). https://doi.org/10.1007/s00706-019-02458-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-019-02458-8