
Abstract
The preparation, structural characterization, luminescence, and catalytic activity of three palladium(II) complexes bearing imino phenoxide ligands are reported. The X-ray studies revealed that the complexes are mononuclear with palladium centres coordinated in a square-planar coordination environment. Two of the complexes are emissive in solution at room temperature. The catalytic activities towards the ring-opening polymerization of rac-lactide (rac-LA) were tested. Polymers with moderate molecular weights and relatively broad dispersity (Ð) were obtained. Kinetic studies revealed that the polymerization followed first-order kinetics.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tetradentate salen-type ligands are omnipresent in coordination chemistry. They are easy to synthesize and they exhibit a high structural variability due to a large number of commercial available building blocks which can be used as starting materials. In many cases, they form highly stable metal complexes throughout the whole periodic table and a plethora of metal complexes have been reported in the past [1, 2]. Related imino phenoxide ligands could be considered as half salen-type ligands and are somewhat less frequently used as ligands. They also exhibit similar advantageous coordination modes, i.e. the combination of a moderate hard and a hard donor atom with an anionic nature of the ligand. Hence, reports from almost all areas of coordination chemistry have been published up to now. Such complexes have found applications in catalysis or biochemistry or feature interesting structural, magnetic, photophysical or electrochemical properties [3,4,5,6,7,8,9,10,11,12].
Besides ubiquitous applications in catalysis for coupling reactions, palladium complexes were intensively investigated due to their interesting luminescence properties [13, 14]. However, only limited number of papers has been published investigating emissive properties using salen-type or imino phenoxide ligands [15]. Palladium in its oxidation state +2 has a d8 configuration and hence prefers a square-planar coordination. It is well known that such complexes form polymeric columnar structures, often with infinite and close Pd–Pd bonds. Contrary to their platinum congeners [16], such complexes usually do not feature luminescence from excited states based on these metallophilic interactions. However, Pd(II) complexes bearing ligands with energetic low-lying π*-accepting orbitals exhibit phosphorescence from either metal-to-ligand charge transfer (3MLCT) or intraligand (3IL) excited states [17].
In recent publications we have shown that main group as well as transition metal imino phenoxide complexes can be used as polymerization catalysts, e.g. for the preparation of polyesters like poly(lactic acid) (PLA) and poly(caprolactone) (PCL) [18,19,20,21,22,23]. They constitute an interesting family of environmentally benign biodegradable polymers with a wide variety of potential applications in the biomedical area or as alternatives to persistent polyolefin materials [24,25,26,27,28,29]. Usually, aliphatic polyesters are prepared by the ring-opening polymerization (ROP) of cyclic esters using metal-based initiators, particularly of toxic tin containing compounds. In the past years, much effort has been spent in studies with alternative catalyst systems containing other, partly less toxic metals [30,31,32,33,34,35,36,37].
In this contribution, we report on the synthesis, structural characterization, photophysical, and catalysis studies of some square-planar Pd(II) complexes bearing imino phenoxide ligands with different structural modifications. Furthermore, although similar Pd(II) complexes have been prepared previously, to the best of our knowledge, the catalysis for the ROP of rac-LA using Pd(II) complexes containing the imino phenoxide backbone is still unreported [38,39,40,41,42,43].
Results and discussion
Synthesis and characterization
The ligands were prepared following a reported literature procedure [44, 45]. The complexes were synthesized by mixing an ethanolic solution of the Schiff base and Pd(II) acetate in refluxing ethanol (Scheme 1).

Complexes 1–3 were isolated in high yields. Their identity and purity are supported by elemental analysis, NMR spectroscopy, and mass spectrometry. In the ESI-MS spectra, signals representing the ions [L2PdH]+ and [L2PdNa]+ are detected. The IR spectra of the free ligands show intense signals in the region from 1623 to 1632 cm−1 (Fig. S10). In the complexes, the spectra are shifted towards lower frequencies (1607–1624 cm−1), which eventually revealed the coordination of imine nitrogen atoms to the metal (Fig. S11). In the 1H NMR spectra, the formation of the metal complexes is shown by the disappearance of the phenol –OH signals of the ligands. In addition, the signals corresponding to the protons of the CH=N groups appear low field shifted (7.52–7.59 ppm) compared to those of the ligands (8.4–8.6 ppm) which indicates the coordination of nitrogen and oxygen atoms of the ligands to the metal.
Structural studies
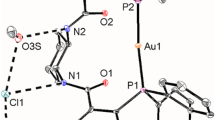
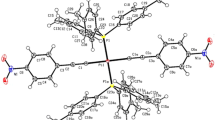
Single crystals suitable for X-ray diffraction of all three complexes were grown by slow evaporation of the solvent from their solutions in dichloromethane over a period of 1 week. All complexes were obtained as red crystalline solids. The crystallographic data are summarized in Table 4. Molecular structures and selected bond lengths and bond angles are displayed in Fig. 1 and Table 1, respectively. Complex 1 crystallizes in the monoclinic space group P21/n, complex 2 in the triclinic space group P \(\bar{1}\), both with one half formula units in the asymmetric unit with the palladium atom as the inversion centre. Complex 2 is isostructural to the homologues copper(II) complex [12]. The palladium atom is coordinated by two nitrogen and two oxygen atoms with an angular sum of exact 180° indicative for a perfect planar coordination. The coordination geometry of 2 is further stabilized by C–Br/π-interactions: the bromine atom points directly to π-system of the phenyl-group of the aniline moiety with a distance between the ring plane and the bromine atom of ~ 3.44 Å which is typical for this kind of interactions [46, 47]. Similarly, for 1 the methyl-substituents are engaged in C–H/π-interactions. 3 is monoclinic, P \(\bar{1}\) with two complexes per asymmetric unit. Contrary to 1 and 2, the palladium atom in 3 exists in a strongly distorted square-planar coordination environment [angular sum 362.0° (Pd1)/361.1° (Pd2)]. This might be due to steric reasons and packing effects. The palladium atoms in all complexes are not engaged in any further Pd–Pd interactions.

Molecular structures of 1–3; thermal ellipsoids were drawn at 50% probability level. Hydrogen atoms were removed for clarity
Electronic spectra
The electronic spectra of all the complexes were recorded in solution at room temperature (Fig. 2). The data are summarized in Table 2. The absorption spectra are very similar to the reported ones [39, 40, 42]. The presence of bands below 300 nm is assigned to π–π* transition of the ligands. The long wavelength absorptions can be assigned to an MLCT. Related Pd(II) complexes bearing salen-type ligands are sometimes only weakly emissive. An recent theoretical study on Pd(II) salen complexes explains the poor emissive behaviour by a low energy gap between emissive MLCT and quenching dd excited states [15]. The authors suggest a substitution of the ligands with strongly electron-withdrawing ligands. Indeed, the ligand in complex 2 carries the moderately electron-withdrawing bromine substituent, whereas the ligand in the non-emissive complex 1 is substituted by a strongly electron-donating methoxy group. For 3, the ligand is substituted with a very bulky t-butyl group which leads not only to a strongly distorted coordination geometry as elucidated by the solid state structure, but also to a more rigid structure with respect to a distortion in the excited state. Minimizing the excited state structural distortion has been found to be effective in increasing the luminescence quantum yield. It should be noted that analogous Pt-complexes often feature intensive luminescence [14].

UV–Vis absorption and emission spectra of 1–3 in DCM at room temperature
Polymerization studies
The complexes 1–3 were investigated as catalysts for ROP of rac-LA (Scheme 2). The kinetics of polymerization was examined by 1H NMR spectroscopy. The polymerization was performed in a 200:1 ratio ([rac-LA]0/[Cat]0 = 200:1) catalysed by complexes 1–3. The plot of % conversion of rac-LA against time gives a sigmoid curve meaning that initially the conversion rate is high but decreases significantly at later stage (Fig. 3, left). There is a linear correlation of the values in the semi-logarithmic plot of ln([rac-LA]0/[rac-LA]t) vs. time indicative for a first-order dependency on the monomer concentrations with the absence of any induction period (Fig. 3, right). This result suggests that the active species remained unchanged and active during the entire course of polymerization. The apparent rate constants (k app) were extracted from the linear plot of ln([rac-LA]0/[rac-LA]t) vs. time and were found to be 3.47 × 10−2 min−1, 2.14 × 10−2 min−1, and 2.84 × 10−2 min−1 for 1–3, respectively (Fig. 2, right), i.e. the activities of the catalysts are slightly higher than those found for the copper(II) homologue complexes we reported previously [12].


rac-LA conversion vs. time (left) and ln ([rac-LA]0/[rac-LA]t) vs. time plot (right) using 1–3: [rac-LA]0:[Cat]0 = 200:1 at 140 °C
Furthermore, bulk polymerizations were performed in a 200:1 (monomer:catalyst) ratio at a temperature of 140 °C and the polymerization was terminated once the rise in the viscosity was observed and the stirring finally ceased. In these experiments, all complexes exhibits a comparable activity to that already found for the kinetic studies above. The representative results are displayed in Table 3. All complexes yielded polymers with moderate molecular weight (M n) and relatively broad dispersities (M w/M n = 1.91–1.98). The large difference between the experimental and theoretic values and broad Ð compared to literature values [48] are thought to be due to the occurrence of polymerization side reactions such as inter-molecular or intra-molecular transesterification as well as a slow initiation rate compared with a fast propagation [49,50,51,52].
Conclusion
In conclusion, we have synthesized three new palladium complexes containing imino phenoxide ligands. The single-crystal X-ray analysis revealed that the palladium atom is surrounded by two ligands in a square-planar coordination environment. Complex 2 and 3 are emissive in solution at room temperature. These compounds have catalytic activity towards the polymerization of rac-LA. The M n of the polymers is moderate with relatively broad M w/M n values.
Experimental
All manipulations were carried out in an atmosphere of dry nitrogen using standard Schlenk techniques. CDCl3 used for NMR spectral measurements was dried over calcium hydride for 48 h, distilled and stored in a glove box. 1H and 13C{1H} NMR spectra were recorded in Bruker Digital Avance III (300 MHz) instrument. Chemical shifts for 1H was referenced to residual solvent resonances and are reported as parts per million relative to SiMe4. IR spectroscopy was performed on a Shimadzu IRAffinity-1 FTIR spectrophotometer that was equipped with a Specac Golden GateTM single-reflection diamond ATR accessory. Mass spectra were collected on a Finnigan LCQ DecaXPPlus ion trap mass spectrometer with an ESI ion source. Elemental analyses were carried out at the Institute for Chemical Technology of Organic Materials at Johannes Kepler University Linz. MALDI-TOF measurements were performed on a Bruker Daltonics instrument in a dihydroxybenzoic acid matrix. For photophysical characterization, spectroscopic grade solvents were used throughout all measurements. Absorption spectra were recorded with a Varian Cary 50 Conc spectrophotometer. All ε values are given in dm3 mol−1 cm−1.
Dichloromethane was dried and distilled over K2CO3, and ethanol was dried and distilled over sodium. Pd(OAc)2 (Sigma-Aldrich) was used without further purification. rac-LA (Sigma-Aldrich) was sublimed under an argon atmosphere repeatedly for further purification and stored in a glove box. All other solvents and reagents were commercially available and used as received. Ligands L1–L3 were prepared according to literature-reported procedures [44, 45]. Polymerization experiments and characterization of the polymers were performed according to described procedures [12].
General procedure for the synthesis of 1–3
The ethanolic solution (10 cm3) of the Schiff base (1 mmol) and the palladium acetate (0.5 mmol) in 10 cm3 ethanol were mixed thoroughly and the mixture was heated under reflux for 2 h and then cooled to room temperature. After filtration the resulting solution was evaporated to dryness and the residue was recrystallized from dichloromethane.
Bis[2-[(4-methoxyphenyl)iminomethyl]-4,6-dimethylphenolato-κ2 N,O 1 ]palladium(II) (1, C32H32N2O4Pd)
Yield: 0.07 g (79%); 1H NMR (300 MHz, CDCl3): δ = 1.26 (s, CH 3, 6H), 2.12 (s, CH 3, 6H), 3.84 (s, Ar–O–CH 3, 6H), 6.76 (s, Ar–H, 2H), 6.83 (s, Ar–H, 2H), 6.90-6.93 (d, J = 9 Hz, Ar–H, 4H), 7.33–7.36 (d, J = 9 Hz, Ar–H, 4H), 7.59 (s, CH=N, 2H) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ = 15.77 (CH3), 20.10 (CH3), 55.88 (Ar–O–CH3), 114.26 (Ar–C), 114.74 (Ar–C), 119.05 (Ar–C), 123.60 (Ar–C), 125.48 (Ar–C), 129.59 (Ar–C), 131.23 (Ar–C), 137.09 (Ar–C), 144.06 (Ar–C), 158.37 (Ar–O), 163.35 (CH=N) ppm; IR (ATR): \(\bar{\nu }\) = 1617 (–CH=N), 1506 (Ar-OMe) cm−1; MS (ESI): m/z calculated for C32H32N2O4PdNa ([M + Na]+) 637.13, found 637.33.
Bis[2,4-dibromo-6-[(2,6-diisopropylphenyl)iminomethyl]phenolato-κ2 N,O 1 ]palladium(II) (2, C38H40Br4N2O2Pd)
Yield: 0.07 g (73%); 1H NMR (300 MHz, CDCl3): δ = 1.17–1.20 (d, J = 9 Hz, CH(CH 3)2, 12H), 1.29–1.32 (d, J = 9 Hz, CH(CH 3)2, 12H), 3.52–3.66 (m, CH(CH3)2, 4H), 7.15–7.16 (d, J = 3 Hz, Ar–H, 2H), 7.18-7.19 (d, J = 3 Hz, Ar–H, 2H), 7.21 (s, Ar–H, 3H), 7.28–7.29 (d, J = 3 Hz, Ar–H, 1H), 7.44 (s, Ar–H, 2H), 7.52–7.53 (d, J = 3 Hz,CH = N, 2H) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ = 23.48 (CH(CH3)2), 24.42 (CH(CH3)2), 28.81 (CH(CH3)2), 105.27 (Ar–C), 114.32 (Ar–C), 120.93 (Ar–C), 124.70 (Ar–C), 127.54 (Ar–C), 135.44 (Ar–C), 140.31 (Ar–C), 142.02 (Ar–C), 145.07 (Ar–C), 159.98 (Ar–O), 163.25 (CH=N) ppm; IR (ATR): \(\bar{\nu }\) = 1607(–CH=N) cm−1; MS (ESI): m/z calculated for C38H41Br4N2O2Pd ([M + H]+) 983.780, found 983.497.
Bis[2-[(4-methoxybenzyl)iminomethyl]-4-methyl-6-(tert-butyl)phenolato-κ2 N,O 1 ]palladium(II) (3, C40H48N2O4Pd)
Yield: 0.06 g (75%); 1H NMR (300 MHz, CDCl3): δ = 1.37 (s, C(CH 3)3, 18H), 2.18 (s, CH 3, 6H), 3.84 (s, Ar–O–CH 3, 6H), 4.98 (s, Ar–CH 2, 4H), 6.79–6.80 (d, J = 3 Hz, Ar–H, 2H), 6.86–6.89 (d, J = 9 Hz, Ar–H, 4H), 7.06–7.07 (d, J = 3 Hz, Ar–H, 2H), 7.48 (s, Ar–H, 2H), 7.53–7.56 (d, J = 9 Hz, CH=N, 2H) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ = 20.40 (CH3), 29.52 (C(CH3)3), 35.22 (C(CH3)3), 55.38 (Ar–O–CH3), 60.87 (Ar–CH2), 114.31 (Ar–C), 122.32 (Ar–C), 123.14 (Ar–C), 129.35 (Ar–C), 130.23 (Ar–C), 131.16 (Ar–C), 131.83 (Ar–C), 133.35 (Ar–C), 139.33 (Ar-C), 159.12 (Ar–O), 165.95 (CH=N) ppm; IR (ATR): \(\bar{\nu }\) = 1624 (–CH=N), 1512 (Ar–OMe) cm−1; MS (ESI): m/z calculated for C40H49N2O4Pd ([M + H]+) 727.27, found 727.60.
X-ray structure determination of compounds 1–3
Suitable single crystals for X-ray diffraction were grown from concentrated dichloromethane solution of the respective compounds over a period of 7 days. Single crystal analysis were carried out on a Bruker SMART APEX and a Bruker Smart X2S diffractometer operating with Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods (SHELXS-97, SIR-97) [53, 54] and refined by full-matrix least squares on F 2 (SHELXL-97) [55]. The H atoms were calculated geometrically, and a riding model was applied in the refinement process. These data were deposited with CCDC with the following numbers: CCDC 1585795–1585797. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk. The crystal data are given in Table 4.
References
Cozzi PG (2004) Chem Soc Rev 33:410
Hernández-Molina R, Mederos A (2003) Acyclic and Macrocyclic Schiff Base Ligands. In: McCleverty JA, Meyer TJ (eds) Comprehensive coordination chemistry II, vol 1. Elsevier, p 411
Li XF, Li YS (2002) J Polym Sci Part A Polym Chem 40:2680
Chellan P, Stringer T, Shokar A, Dornbush PJ, Vazquez-Anaya G, Land KM, Chibale K, Smith GS (2011) J Inorg Biochem 105:1562
Cui J, Zhang M, Zhang Y (2010) Inorg Chem Commun 13:81
Younkin TR, Connor EF, Henderson JI, Friedrich SK, Grubbs RH, Bansleben DA (2000) Science 287:460
Henderson W, Evans C, Nicholson BK, Fawcett J (2003) Dalton Trans:2691
Zheng F, Hutton AT, van Sittert CGCE, Moss JR, Mapolie SF (2013) Dalton Trans 42:11163
Chellan P, Shunmoogam-Gounden N, Hendricks DT, Gut J, Rosenthal PJ, Lategan C, Smith PJ, Chibale K, Smith GS (2010) Eur J Inorg Chem:3520
Komiya N, Okada M, Fukumoto K, Jomori D, Naota T (2011) J Am Chem Soc 133:6493
Komiya N, Muraoka T, Iida M, Miyanaga M, Takahashi K, Naota T (2011) J Am Chem Soc 133:16054
Mandal M, Oppelt K, List M, Teasdale I, Chakraborty D, Monkowius U (2016) Monatsh Chem 147:1883
Xu G, Luo Q, Eibauer S, Rausch RF, Stempfhuber S, Zabel M, Yersin H, Reiser O (2011) Dalton Trans 40:8800
Chow PK, Cheng G, Tong GSM, Ma C, Kwok WM, Ang WH, Chung CYS, Yang C, Wang F, Che CM (2016) Chem Sci 7:6083
Tong GSM, Chow PK, To WP, Kwok WM, Che CM (2014) Chem Eur J 20:6433
Williams JAG (2007) Top Curr Chem 281:205
Yersin H, Rausch AF, Czerwieniec R, Hofbeck T, Fischer T (2011) Coord Chem Rev 255:2622
Saha TK, Mandal M, Chakraborty D, Ramkumar V (2013) New J Chem 37:949
Mandal M, Monkowius U, Chakraborty D (2016) New J Chem 40:9824
Chakraborty D, Chokkapu ER, Mandal M, Gowda RR, Ramkumar V (2016) ChemistrySelect 1:5218
Rajashekhar B, Mandal M, Chakraborty D, Ramkumar V (2017) ChemistrySelect 2:8408
Mandal M, Monkowius U, Chakraborty D (2016) J Polym Res 23:220
Saha TK, Mandal M, Thunga M, Ramkumar V, Chakraborty D (2013) Dalton Trans 42:10304
Ragauskas AJ, Williams CK, Davison BH, Tschaplinski T (2006) Science 311:484
Williams CK, Hillmyer MA (2008) Polym Rev 48:1
Dove AP (2008) Chem Commun 48:6446
Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P (2013) Chem Soc Rev 42:1147
Dechy-Cabaret O, Martin-Vaca B, Bourissou D (2004) Chem Rev 104:6147
Jerome C, Lecomte P (2008) Adv Drug Delivery Rev 60:1056
Mandal M, Chakraborty D (2016) J Polym Sci Part A Polym Chem 54:809
Mandal M, Chakraborty D, Ramkumar V (2015) RSC Adv 5:28536
Tsai C-Y, Du H-C, Chang J-C, Huang B-H, Ko B-T, Lin C-C (2014) RSC Adv 4:14527
Wang L, Poirier V, Ghiotto F, Bochmann M, Cannon RD, Carpentier J-F, Sarazin Y (2014) Macromolecules 47:2574
Aluthge DC, Patrick BO, Mehrkhodavandi P (2013) Chem Commun 49:4295
Sauer A, Kapelski A, Fliedel C, Dagorne S, Kol M, Okuda J (2013) Dalton Trans 42:9007
Dagorne S, Normand M, Kirillov E, Carpentier J-F (2013) Coord Chem Rev 257:1869
Nie K, Gu W, Yao Y, Zhang Y, Shen Q (2013) Organometallics 32:2608
Joshi H, Prakash O, Sharma AK, Sharma KN, Singh AK (2015) Eur J Inorg Chem:1542
Blackburn OA, Coe BJ, Fielden J, Helliwell M, McDouall JJW, Hutchings MG (2010) Inorg Chem 49:9136
Roy S, Saha R, Mondal TK, Sinha C (2014) Inorg Chim Acta 423:52
Feng ZQ, Yang XL, Ye YF, Hao LY (2014) Bull Korean Chem Soc 35:1121
Khorshidifard M, Rudbari HA, Askari B, Sahihi M, Farsani MR, Jalilian F, Bruno G (2015) Polyhedron 95:1
Tajuddin AM, Bahron H, Zaki HM, Kassim K, Chantrapromma S (2015) Acta Crystallogr E71:350
Kasumov VT, Köksal F, Sezer A (2005) Polyhedron 24:1203
Bhunora S, Mugo J, Bhaw-Luximon A, Mapolie S, Wyk JV, Darkwa J, Nordlander E (2011) Appl Organomet Chem 25:133
Swierczynski D, Luboradzki R, Dolgonos G, Lipkowski J, Schneider H-J (2005) Eur J Org Chem:1172
Nagels N, Hauchecorne D, Herrebout WA (2013) Molecules 18:6829
Masutani K, Yoshiharu K (2015) PLA Synthesis and Polymerization. In: Jiménez A, Peltzer M, Ruseckaite R (eds), Poly(lactic acid) science and technology: processing, properties, additives and applications. RSC polymer chemistry series no. 12, The Royal Society of Chemistry, p 3
Kricheldorf HR, Mang T, Jonte JM (1984) Macromolecules 17:2173
Dubois P, Jacobs C, Jérôme R, Teyssié P (1991) Macromolecules 24:2266
Stevels WM, Ankone MJ, Dijkstra PJ, Feijen J (1996) Macromolecules 29:6132
Chamberlain BM, Jazdzewski BA, Pink M, Hillmyer MA, Tolman WB (2000) Macromolecules 33:3970
Sheldrick GM (1997) SHELXS-97, program for the solution of crystal structures. Göttingen, Germany. See also: Sheldrick GM (1990). Acta Crystallogr A46:467
Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R (1999) J Appl Cryst 32:115
Sheldrick GM (1997) SHELXL-97, program for crystal structure refinement. Göttingen, Germany. See also Sheldrick GM (2008). Acta Crystallogr A64:112
Acknowledgements
Open access funding provided by Johannes Kepler University Linz. MM thanks for a scholarship within the framework of Erasmus Mundus Action 2 doctoral exchange program of the European Commission. We thank the JKU and Prof. Knör (JKU) for his generous support of the experimental work. The NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mandal, M., List, M., Teasdale, I. et al. Palladium complexes containing imino phenoxide ligands: synthesis, luminescence, and their use as catalysts for the ring-opening polymerization of rac-lactide. Monatsh Chem 149, 783–790 (2018). https://doi.org/10.1007/s00706-017-2119-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-017-2119-1