
Abstract
The tetrazine ligation is one of the fastest bioorthogonal ligations and plays a pivotal role in time-critical in vitro and in vivo applications. However, prediction of the reactivity of tetrazines in inverse electron demand Diels–Alder-initiated ligation reactions is not straight-forward. Commonly used tools such as frontier molecular orbital theory only give qualitative and often even wrong results. Applying density functional theory, we have been able to develop a simple computational method for the prediction of the reactivity of aryl/alkyl-substituted tetrazines in inverse electron demand Diels–Alder reactions.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tetrazine ligations (TLs) are bioorthogonal inverse electron demand Diels–Alder (IEDDA) initiated cycloadditions proceeding with exceptional high second-order rates of up to 3,300,000 M−1 s−1 [1]. In TLs, an 1,2,4,5-tetrazine (Tz) reacts with an electron-rich dienophile in an IEDDA reaction followed by cycloreversion under the loss of nitrogen (Fig. 1). Strained alkenes such as norbornenes [2, 3], cyclopropenes [4, 5], and trans-cyclooctenes (TCOs) [1, 6,7,8] are commonly used dienophiles, with TCOs providing the highest reactivity. The rate-determining step is the Diels–Alder cycloaddition, while the cycloreversion has only a low energy barrier and is suspected to show strong non-statistical effects [9].

Mechanism of the bioorthogonal ligation of 1,2,4,5-tetrazine (Tz) and trans-cyclooctene (TCO)
Due to the high reaction rates, these ligations can be used in time-critical applications such as rapid radiolabeling and pretargeted PET imaging [10,11,12,13,14,15,16] and provide high yields within short reaction times even at low concentrations as usually encountered in radiochemistry and in vivo. Therefore, kinetics is one of the most important characteristics of bioorthogonal reactions. However, prediction of reactivities using the chemist’s understanding of organic chemistry, especially of IEDDA reactions, might lead to wrong predictions [17] and only qualitative estimates. In addition, synthesis of tetrazines is often low yielding and involves handling or even requires the production of anhydrous hydrazine (not commercially available in Europe), which limits the feasibility of screening for high Diels–Alder reactivity. Hence, there is the need of reliable computational tools to predict the reactivity of various tetrazines in TLs.
Herein, we introduce a computational model for the prediction of the reactivity of aryl/alkyl-substituted Tz in the cycloaddition with trans-cyclooctene (TCO), thus eliminating the need for expensive and even dangerous synthetic work, finally enabling in silico screening for tetrazines with desired reactivity.
While 3,6-bisaryl- and 3-aryl-substituted Tz show the highest reactivity, aryl/alkyl-substituted Tz are commonly used due to higher stability [18, 19] and show favorable properties in Tz-triggered bioorthogonal elimination reactions [20, 21].
Results and discussion
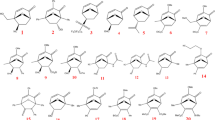
Recently, we have investigated the reactivity of several 3-aryl-6-(3-fluoropropyl)-1,2,4,5-tetrazines 1–8 as chemical probes for rapid radiolabeling and pretargeted PET imaging (Fig. 2). While the alkyl substituent is the same for all eight tetrazines the aryl component shows considerable variation including electron-rich and electron-poor aryl groups. In addition, Tz 9 and 10 were included to investigate the influence of the alkyl group and an ortho-substituted aryl residue, respectively.

Investigated aryl/alkyl substituted 1,2,4,5-tetrazines in the IEDDA reaction with trans-cyclooctene (11)
The second-order rate constants of Tz 1–10 in the reaction with trans-cyclooctene 11 at 25 °C in anhydrous 1,4-dioxane were measured by stopped-flow spectrophotometry, which gave rates ranging from 1.00 M−1 s−1 for electron-rich trimethoxyphenyl-substituted Tz 3 to 14.6 M−1 s−1 for Tz 8 bearing an electron withdrawing 2-pyridyl substituent (Fig. 3).

Second-order rate constants of tetrazines 1–10 with trans-cyclooctene (11) in 1,4-dioxane at 25 °C
These experimental results were selected as a basis for the construction of a predictive computational tool. DFT was successfully used in the past by our group [16, 22] and others [7, 17, 23, 24] to predict or explain the reactivity of dienophiles and tetrazines in the tetrazine ligation. Therefore, the Minnesota density functional M06-2X in combination with the 6-311+G(d,p) basis set, was used as model chemistry. This density functional has been proven to produce accurate results for thermodynamics of cycloaddition reactions [25, 26].
Diels–Alder reactions can be described by HOMO/LUMO interactions using the frontier molecular orbital (FMO) theory. In case of the IEDDA cycloaddition the main orbital interaction is between a low-lying unoccupied orbital of the dienophile, usually being the LUMO+1 for aryl/alkyl tetrazines (Fig. 4a) [17], and the HOMO of the electron-rich dienophile (in this case TCO, Fig. 4b).

a HOMO, LUMO, and LUMO+1 of 3-methyl-6-phenyl-Tz (9); b HOMO of TCO (11); c energy levels of selected orbitals for tetrazines 1–10 and TCO (11)
According to the FMO theory, a smaller energy gap between the interacting orbitals facilitates the reaction. Thus, one might expect that a more electron withdrawing and thus LUMO+1-lowering substituent accelerates the reaction, while an electron-rich aryl substituent will decrease reactivity. HF/6-311+G(d,p)//M06-2X/6-311+G(d,p)-calculated orbital energies are shown in Fig. 4c. Tz 4 and 7 bearing an electron-withdrawing trifluoromethyl or sulfone group, respectively, show the lowest LUMO and LUMO+1 energies. However, the tetrazine with the highest reactivity, Tz 8, has one of the highest LUMO and a rather high LUMO+1 energy within the series. As shown in Fig. 5, there is no significant correlation between the LUMO+1-energy levels and the rate constants (R 2 = 0.07). This can be rationalized by the fact that FMO interactions are not the only major contributors to activation energies in such cycloadditions [17, 27,28,29]. Therefore, the FMO theory cannot be applied for the reliable prediction of the Diels–Alder reactivity of aryl/alkyl-substituted Tz.

Second-order rate constants plotted against HF/6-311+G(d,p)//M06-2X/6-311+G(d,p) calculated LUMO+1 energies of the corresponding tetrazine
However, the calculated energies of activation (ΔE ‡) of the reactions between tetrazines 1–10 and TCO (11) show excellent linear correlation with the natural logarithm of the measured rate constants, as per transition state theory (Fig. 6). M06-2X/6-311+G(d,p) calculated energies of activation can, therefore, be used to reliably predict the reactivity of aryl/alkyl-Tz in IEDDA reactions with trans-cyclooctenes.

Correlation between natural logarithm of second-order rate constants and M06-2X/6-311+G(d,p) calculated energies of activation
Using the linear correlation between ln(k) and calculated ΔE ‡, Eq. (1) can be constructed which allows the prediction of the rate constant of new aryl/alkyl-Tz for the reaction with TCO (11) in anhydrous 1,4-dioxane at 25 °C.
Conclusion
Prediction of the reactivity of Tz in bioorthogonal ligation reactions is essential to reduce synthetic work, the resulting costs and associated risks. However, the chemist’s prediction of these reactivities, mainly based on frontier molecular orbital theory, can only yield qualitative or even wrong results as shown in this work. We were able to develop a computational method for the prediction of reactivities of aryl/alkyl-substituted 1,2,4,5-tetrazines in the reaction with trans-cyclooctene based on M06-2X-calculated energies of activation. This method is computationally cheap as it requires only the optimization of the tetrazine and the corresponding transition state with trans-cyclooctene at the M06-2X/6-311+G(d,p) level of theory, which can even be done on an average desktop PC within hours.
We are convinced that our method will aid the development of new aryl/alkyl tetrazines for bioorthogonal applications, and represents a step forward to the development of a universal computational tool being able to predict the reactivity of tetrazines with various substitution patterns.
Experimental
Computational methods
Calculations were performed at the M06-2X/6-311+G(d,p) level of theory in the gas phase using the Gaussian 09 Rev. D.01 software package [30]. Vibrational analysis was performed to confirm stationary points are energetic minima or transition states, respectively. Orbital energies were calculated by performing a HF/6-311+G(d,p) single-point calculation on M06-2X/6-311+G(d,p) optimized structures. ΔE ‡ was determined by calculating the difference in energies between transition state and reactants. Data analysis was preformed using Gaussview 5 and Chemcraft. XYZ coordinate files of all reactants and transition states are available as electronic supplementary material. Calculated energies for Tz 1–10 and the respective transition states (for the reaction with TCO) are shown in Table 1.
Synthesis
Trans-cyclooctene (11) and tetrazines 9 and 10 were prepared following known procedures [31, 32]. The synthesis of compounds 1–8 will be published elsewhere (manuscript in preparation).
Kinetic measurements
Kinetic measurements were performed on a SX20 stopped-flow spectrophotometer (Applied Photophysics, UK) using a 535 nm LED light source. A 4 mM solution of 11 and approximately 0.1 mM solutions of Tz 1–10 were prepared in anhydrous 1,4-dioxane (note: for correct measurements it is of utmost importance to use anhydrous 1,4-dioxane, since even small amounts of water will accelerate the reaction, leading to irreproducible data). These solutions were loaded into the driver syringes and equal volumes of TCO and Tz solution were mixed, resulting in concentrations of 2 and 0.05 mM, respectively. The reaction progress was followed by measuring the absorption around 535 nm. All measurements were performed in triplicates. Observed reaction constants (k obs) were determined by non-linear regression (one-phase-decay) using Prism 6 (Graphpad) and second-order rate constants (k) were calculated from k obs. Table 2 lists k obs, c TCO, c Tz and k for all reactions.
References
Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM (2014) Chem Sci 5:3770
Devaraj NK, Weissleder R, Hilderbrand SA (2008) Bioconjug Chem 19:2297
Knall A-C, Hollauf M, Slugovc C (2014) Tetrahedron Lett 55:4763
Yang J, Šečkutė J, Cole CM, Devaraj NK (2012) Angew Chem Int Ed 51:7476
Yang J, Liang Y, Šečkutė J, Houk KN, Devaraj NK (2014) Chemistry 20:3365
Rossin R, van den Bosch SM, Ten Hoeve W, Carvelli M, Versteegen RM, Lub J, Robillard MS (2013) Bioconjug Chem 24:1210
Taylor MT, Blackman ML, Dmitrenko O, Fox JM (2011) J Am Chem Soc 133:9646
Selvaraj R, Fox JM (2013) Curr Opin Chem Biol 17:753
Törk L, Jiménez-Osés G, Doubleday C, Liu F, Houk KN (2015) J Am Chem Soc 137:4749
Wang M, Svatunek D, Rohlfing K, Liu Y, Wang H, Giglio B, Yuan H, Wu Z, Li Z, Fox J (2016) Theranostics 6:887
Wu Z, Liu S, Hassink M, Nair I, Park R, Li L, Todorov I, Fox JM, Li Z, Shively JE, Conti PS, Kandeel F (2013) J Nucl Med 54:244
Rossin R, van Duijnhoven SMJ, van den Bosch SM, Robillard MS (2014) Nucl Med Biol 41:630
Li Z, Cai H, Hassink M, Blackman ML, Brown RCD, Conti PS, Fox JM (2010) Chem Commun 46:8043
Liu S, Hassink M, Selvaraj R, Yap L, Park R, Wang H, Chen X, Fox JM, Li Z, Conti PS (2013) Mol Imaging 12:121
Meyer JP, Houghton JL, Kozlowski P, Abdel-Atti D, Reiner T, Pillarsetty NVK, Scholz WW, Zeglis BM, Lewis JS (2016) Bioconjug Chem 27:298
Denk C, Svatunek D, Filip T, Wanek T, Lumpi D, Fröhlich J, Kuntner C, Mikula H (2014) Angew Chem Int Ed 53:9655
Liu F, Liang Y, Houk KN (2014) J Am Chem Soc 136:11483
Carlson JCT, Meimetis LG, Hilderbrand SA, Weissleder R (2013) Angew Chem Int Ed 52:6917
Karver MR, Weissleder R, Hilderbrand SA (2011) Bioconjug Chem 22:2263
Fan X, Ge Y, Lin F, Yang Y, Zhang G, Ngai WSC, Lin Z, Zheng S, Wang J, Zhao J, Li J, Chen PR (2016) Angew Chem Int Ed 55:14046
Rossin R, van Duijnhoven SMJ, ten Hoeve W, Janssen HM, Kleijn LHJ, Hoeben FJM, Versteegen RM, Robillard MS (2016) Bioconjug Chem 27:1697
Denk C, Svatunek D, Mairinger S, Stanek J, Filip T, Matscheko D, Kuntner C, Wanek T, Mikula H (2016) Bioconjug Chem 27:1707
Liang Y, Mackey JL, Lopez S, Liu F, Houk KN (2012) J Am Chem Soc 134:17904
Lambert WD, Scinto SL, Dmitrenko O, Boyd SJ, Magboo R, Mehl RA, Chin JW, Fox JM, Wallace S (2017) Org Biomol Chem 15:6640
Lan Y, Zou L, Cao Y, Houk KN (2011) J Phys Chem A 115:13906
Paton RS, Mackey JL, Kim WH, Lee JH, Danishefsky SJ, Houk KN (2010) J Am Chem Soc 132:9335
Levandowski BJ, Houk KN (2015) J Org Chem 80:3530
Liu F, Paton RS, Kim S, Liang Y, Houk KN (2013) J Am Chem Soc 135:15642
Levandowski BJ, Hamlin TA, Bickelhaupt FM, Houk KN (2017) J Org Chem 82:8668
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 09, Rev. D.01. Gaussian Inc., Wallingford
Svatunek D, Denk C, Rosecker V, Sohr B, Hametner C, Allmaier G, Fröhlich J, Mikula H (2016) Monatsh Chem 147:579
Suh S-E, Barros SA, Chenoweth DM (2015) Chem Sci 6:5128
Acknowledgements
Open access funding provided by TU Wien (TUW). The authors thank Heidi Fittner (TU Wien) for her support. Quantum-chemical calculations were performed using the Vienna Scientific Cluster (VSC). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreement No 668532 (‘Click-It’).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Svatunek, D., Denk, C. & Mikula, H. A computational model to predict the Diels–Alder reactivity of aryl/alkyl-substituted tetrazines. Monatsh Chem 149, 833–837 (2018). https://doi.org/10.1007/s00706-017-2110-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-017-2110-x