
Abstract
A high yield preparation, spectroscopic and crystallographic investigation of the crystalline Zn-complex of a di(β,β′-sulfoleno)pyrrin are reported here. In the brightly green fluorescent Zn-complex of the hardly luminescent di(β,β′-sulfoleno)pyrrin, the metal ion is bound by two di(β,β′-sulfoleno)pyrrin ligands, as revealed first by its mass spectra. The crystal structure of this Zn-complex of the di(β,β′-sulfoleno)pyrrin confirmed a regular 2:1 composition of the bidentate di(β,β′-sulfoleno)pyrrin ligand and the metal ion. The latter was coordinated in a distorted tetrahedral fashion, as found in other dipyrrin Zn-complexes. The here studied Zn-complex of a designed di(β,β′-sulfoleno)pyrrin ligand provides insights into the coordination properties of the proposed (2:1)- and (2:2)-complexes of phylloxanthobilin and bilirubin, respectively, which are two abundant natural bilin-type tetrapyrroles.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dipyrrins (or dipyrromethenes) feature two conjugated pyrrole rings and represent (formal) dipyrrolic building blocks for the construction of porphyrins and related tetrapyrrolic macrocycles [1]. The complexes of the bidentate dipyrrins with boron (the ‘BODIPY’-complexes) [2] or with transition metal ions [3, 4] have attracted particular attention due to the ‘predictable’ coordination properties of dipyrrins, and the tunable emission and absorption properties of their metal complexes [5–9]. Hence, the design of dipyrrin ligands has been attractive, and dipyrrin chemistry has taken advantage of the development of a broad range of strategies for their construction [2–5, 7, 8]. In one approach (used here), dipyrrins are prepared by dehydrogenation of easily accessible corresponding dipyrromethanes [3, 4].
In the context of our recent interest in the metal coordination properties of yellow chlorophyll catabolites (phylloxanthobilins) [10], and of other natural linear tetrapyrroles derived from chlorophyll [11, 12], we report here on our investigations of a model dipyrrole, the di(β,β′-sulfoleno)pyrrin 2.
Results and discussion
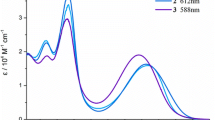
Our synthetic route to di(β,β′-sulfoleno)pyrrin 2 relied on the earlier made corresponding di(β,β′-sulfoleno)pyrromethane (1), available, in turn, from condensation of 3,5-di-tert-butylbenzaldehyde and β,β′-sulfolenopyrrole [13]. Dipyrromethane 1 was oxidized with dicyanodichlorobenzoquinone (DDQ) to furnish bright yellow 2 in 76 % yield, after crystallization in CH2Cl2/n-C6H14 (see Scheme 1). The UV/Vis spectrum of the dipyrrin 2, displayed in Fig. 1, exhibits characteristic maxima at 436.5 and 327 nm, comparable to the one of bilirubin [14, 15], or of a recently described yellow chlorophyll catabolite (YCC, a phylloxanthobilin) [16, 17]. A FAB-mass spectrum featured a strong pseudo-molecular ion at m/z = 513.1 [M + H]+, confirming its molecular formula as C27H32N2O4S2. Fragments at m/z = 448.2 and 384.2 indicated consecutive loss of the two SO2-groups. The 1H NMR spectrum of 2 exhibited two singlets at intermediate field of the two pairs of symmetry equivalent β-methylene groups, a singlet at 7.58 ppm of the pyrrole-α positions, the signals of aryl o- and p-protons at low field, and a broad signal of an NH at 12.76 ppm (see Fig. 2, bottom).


UV–Vis spectra of the dipyrrin 2 and of its Zn-complex 3 (=Zn-(2)2) in CH2Cl2 (4 × 10−6 mol/dm3)

1H NMR spectra of the dipyrrin 2 (bottom) and of its Zn(II)-complex 3 (Zn-(2)2) (top) in CDCl3 (300 MHz, 25 °C, × = solvent signals)
For the preparation of 3, a solution of 15 mg of Zn(OAc)2 2H2O (68.4 μmol, 18 eq) in 0.3 cm3 MeOH was mixed into a solution of 2 mg 2 (3.8 μmol) in 2.7 cm3 of CH2Cl2 at room temperature. After 5 min, the reaction mixture was worked up by extraction and evaporation of the solvent (see “Experimental” part). The Zn-complex 3 crystallized from CH2Cl2/n-C6H14 as pink-red crystals (2.0 mg, 94 % yield).
The molecular formula of the dipyrrin Zn-complex 3 was indicated as C54H62N4O8S4Zn from analysis of its pseudo-molecular ion [M+H]+ at m/z = 1087.2. Corresponding significant fragments occurred at m/z = 1023.3, 894.4, and 830.4, due to consecutive loss of the one, three, and four SO2-groups, respectively. The derived molecular formula of the Zn-complex 3 suggested the presence of two dipyrrin ligands 2 and one Zn(II)-ion, i.e. to represent Zn-(2)2 (see Scheme 2).

The pink-red Zn-complex 3 displayed a UV/Vis-spectrum in CH2Cl2 that featured a maximum at 487 nm (and a shoulder at 463 nm), corresponding to a 51 nm bathochromic shift, when compared with the spectrum of the dipyrrin 2 (see above). Similar bathochromic shifts of the absorption spectrum upon coordination of a Zn(II) ion were observed in Zn-complexes of bilirubin [18], or of phylloxanthobilins [10, 12]. Analysis of the Zn-complex 3 by fluorescence spectroscopy showed an intense emission band at 505 nm, whereas the metal-free dipyrrin 2 displayed little luminescence with a maximum around 530 nm (see Fig. 3). The excitation spectrum of dipyrrin Zn-complex 3, observed at 505 nm, fitted the absorption spectrum of 3. As, in contrast, the dipyrrin 2 was essentially non-luminescent in CH2Cl2, the rapid coordination of Zn ions and formation of 3 lightened up an intense green luminescence with about 200–300-fold higher intensity (see Fig. 3).

Fluorescence spectra of 2 and 3 in CH2Cl2 (top: 2, 4 × 10−6 mol/dm3, EM: excited at 436 nm, EX: observed for 530 nm; bottom: 3, 4 × 10−6 mol/dm3, EM: excited at 487 nm, EX: observed for 505 nm)
In the 1H NMR spectrum (in CDCl3, see Fig. 2) of the Zn-complex 3 [Zn-(2)2], a signal of an HN was lacking (which was found at 12.76 ppm in the spectrum of the dipyrrin 2), consistent with bidentate binding of the dipyrrin ligand 2 to the coordinated Zn(II) ion. Signals of aryl-o hydrogens and aryl-p hydrogen are slightly shifted to lower field while signals of β-methylene groups and pyrrole-α hydrogens are slightly moved to higher field.
X-ray diffraction quality crystals of the Zn-complex 3 grew from a solution of 3 in CH2Cl2 when n-C6H14 was mixed in slowly at 4 °C. The Zn-complex 3 crystalized in the triclinic system with space group P-1 (no. 2). A unit-cell contained four molecules of 3. The crystal structure of 3 showed two bidentate dipyrrin moieties wrapped around one Zn(II) center leading to coordination in a distorted tetrahedral fashion with N–Zn–N angles of about 94, 113, and 121° (Fig. 4). The bonds of the four N atoms to the coordinated Zn ion are 1.98 Å long, consistent with crystallographic data from other Zn-dipyrrin complexes (1.96–1.98 Å) [19–21]. The structure of 3 in the triclinic crystal deviates slightly from the symmetric model reported for crystals of other bis(dipyrrinato) Zn-complexes [19]. The planes of the two dipyrrin ligands are roughly vertical to each other (82° dihedral angle), as are the aryl groups at the 5-position with respect to the conjugated pyrrole system in the same ligand moiety (85°). Thus, the plane defined by one aryl group at the meso-position is observed at 3.8° with respect to the plane of the conjugated pyrrole system in the other ligand. In contrast, the other aryl group at 5-position is inclined by 17.1° with respect to the conjugated pyrrole system in the second dipyrrin unit. Probably, these small symmetry-deviations are consequences of the packing in the triclinic crystal. The bond lengths and bond angles within the dipyrrin ligands in 3 are similar to those of other bis(dipyrrinato) Zn-complexes [19–21]. The crystal structure of 3 reflects the symmetric, bidentate coordination behavior of the dipyrrin 2. Thus, the Zn-complex 3 may represent a valuable model for the chelation pattern in non-crystalline Zn(II)-complexes of natural oligopyrroles with similar chromophores, e.g. of bilirubin [18] or of the phylloxanthobilin YCC [10].

ORTEP-plots of the crystal structure of the bis(dipyrrinato) Zn-complex 3. a Model of the structure of 3 highlighting the two planes spanned by the dipyrrin ligands (H atoms and tert-butyl substituents at meso-aryl groups are omitted). b Model of the distorted tetrahedral core of 3, with specified lengths Zn-N bonds and distances between the N’s (left part) and N–Zn–N bond angles of the Zn–N4 core (right part). c Bond lengths (left) and bond angles (right) in the dipyrrin cores of 3
Conclusion
Dipyrrin 2 is a yellow dipyrrole that possesses two conjugated pyrrolic rings and shows negligible luminescence. It may be considered a simple model compound for the chromophore part of some natural tetrapyrroles, such as the heme-derived bilirubin (BR) [14, 15] and phylloxanthobilins [17] or yellow chlorophyll catabolites (YCCs) [10, 16]. Binding of Zn(II) ions to the bidentate 2 furnishes the 2:1 complex 3, the crystal structure of which exhibited a distorted tetrahedral structure. This type of coordination pattern was derived for the (2:1)-complex of a YCC with Zn(II) ions [10]. Similar, furthermore, to observation with the Zn(II)-complex of the YCC [10], the bis(dipyrrinato) Zn-complex 3 displays intensive green luminescence. Hence, the present study helps to model the coordination behavior of Zn-complexes of natural oligopyrroles with similar conjugated chromophores, e.g. of BR [18] or of YCC [10, 12], and to gain basic insights into their luminescence properties. Bis(dipyrrinato) Zn-complexes, and related boron complexes of dipyrrins (BODIPYs) [2], exhibit intensive and tunable absorption and emission properties, which make them useful in various optical applications [5–7, 9]. In contrast to BODIPYs, bis(dipyrrinato) Zn-complexes exhibit a tetrahedral coordination pattern in 2:1 assemblies (ligand: Zn), giving them considerable potential in supramolecular structuring [21, 22]. The sulfoleno-units of the dipyrrin 2 and of the bis(dipyrrinato) Zn-complex 3 are, furthermore, ‘programmed’ for introduction of covalent modifications at the pyrrole β-positions by [4+2]-cycloaddition reactions. As was recently developed with porphyrinoids, such as tetra-sulfolenoporphyrins [23–25] and a tetra-sulfolenocorrole [13], the dipyrrinato-units of 2 and 3 could, thus, also open up efficient access to further designed functionalized supramolecular assemblies.
Experimental
Dichlorodicyano-p-benzoquinone (DDQ) and zinc acetate dihydrate [Zn(OAc)2 2H2O] were reagent grade commercial chemicals from Fluka and were used as received; EtOAc, dichloromethane, methanol (MeOH), and n-C6H14 were from Acros and were distilled before use. Column chromatography (CC): Fluka silica gel 60 (230–400 mesh). Thin layer chromatography (TLC): Merck 0.25 mm silica gel 60 plates. Equipment: UV/Vis: Agilent Cary 60 UV–Visible, λ max in nm (log ε). Fluorescence (FL): Varian Cary Eclipse, λ in nm (rel. intensity); Nuclear magnetic resonance (1H) spectra: Bruker 300 at 298K, chemical shifts (δ) in ppm, with δ (CHCl3) = 7.26 ppm, signal assignment follows the X-ray numbering scheme. FAB-MS: Finnigan MAT-95, positive ion mode, NOBA matrix; X-ray analyses: data collection on a Nonius Kappa CCD, equipped with graphite mono-chromatized Mo-Kα-radiation (λ = 0.71073 Å) at 233K. Melting point: Büchi 535.
5-(3,5-Di-tert-butylphenyl)-di(β,β′-sulfoleno)pyrrin (2, C27H32N2O4S2)
To the solution of 2 mg dipyrromethane 1 (3.8 μmol) [13] in 1 cm3 CH2Cl2 1.4 mg DDQ (6.2 μmol, 1.6 equiv) was added. After 20 h at room temperature, the reaction mixture was diluted to 20 cm3 with CH2Cl2 and washed with saturated aq. NaHCO3 (3 × 15 cm3). The organic phase was filtered through a plug of dry cotton-wool and evaporated to dryness under reduced pressure to give a brown residue. The residue was dissolved in 1.5 cm3 CH2Cl2 and loaded on a silica gel column (1.5 × 10 cm). The product was washed down with CH2Cl2/EtOAc 10/1 (v/v). The collected product fractions were combined and concentrated to dryness under reduced pressure to furnish 2 as a yellow residue. Dipyrrin 2 was isolated after crystallization from CH2Cl2/n-C6H14 (1/4 cm3) at 4 °C as 1.5 mg of yellow crystals (yield 76 %). M.p.: 178 °C; UV/Vis (CH2Cl2): λ max (log ε) = 327 (3.72), 436.5 (4.42) nm; fluorescence emission (CH2Cl2, c = 4 × 10−6 mol/dm3, excited at 436 nm, rel. intensity): 530 nm (1.00); fluorescence excitation (obs. for 530 nm, rel. intensity): 437 (1.00), 330 (0.40) nm; 1H NMR (300 MHz, CDCl3): δ = 1.35 (s, CH3 of t-Bu), 3.17 (s, H2C31, H2C71), 4.10 (s, H2C21, H2C81), 7.06 (d, J = 1.7 Hz, HC52, HC52′), 7.58 (s, HC1, HC9), 7.61 (t, J = 1.7 Hz, HC54), 12.76 (br s, HN11) ppm; FAB-MS: m/z (%) = 515.2 (14), 514.2 (33), 513.1 (100, [M+H]+, calcd. for C27H32N2O4S2 (512.18), 450.1 (10), 449.2 (27), 448.2 (32, [M–SO2]+), 386.2 (15), 385.2 (52), 384.2 (75, [M–2SO2]+).
Bis(5-(3,5-di-tert-butylphenyl)-di(β,β′-sulfoleno)pyrrinato) Zn-complex (3, C54H62N4O8S4Zn)
Dipyrrin 2 (2 mg, 3.8 μmol) was dissolved in 2.7 cm3 CH2Cl2 and the solution of 15 mg Zn(OAc)2 2H2O (68.4 μmol, 18 eq.) in 0.3 cm3 MeOH was added. After 5 min, the reaction mixture was diluted to 15 cm3 with CH2Cl2 and washed with saturated aq. NaHCO3 (3 × 10 cm3). The organic layer was filter through a plug of dry cotton wool and evaporated to dryness under reduced pressure to give a red residue. After crystallization in CH2Cl2/n-C6H14 (1.5/4 cm3) at 4 °C, 2 mg of 3 were obtained as pink-red crystals, yield is 94 %. M.p.: >195 °C (decomp.); UV/Vis (CH2Cl2): λ max (log ε) = 242 (4.07), 357.5 (3.81), 385sh (3.66), 463sh (4.51), 487 (4.82) nm; fluorescence emission (CH2Cl2, c = 4 × 10−6 mol/dm3, excited at 487 nm, rel. intensity): 505 nm (1.00); fluorescence excitation (obs. for 505 nm, rel. intensity): 486 (1.00), 461sh (0.49), 355 (0.10) nm; 1H NMR (300 MHz, CDCl3): δ = 1.40 (s, CH3 of t-Bu), 3.11 (s, H2C31, H2C71), 4.09 (s, H2C21, H2C81), 7.11 (d, J = 1.7 Hz, HC52, HC52′), 7.54 (s, HC1, HC9), 7.66 (t, J = 1.7 Hz, HC54) ppm (due to the symmetric structure, only half molecular chemical shifts were labeled here); FAB-MS: m/z = 1087.2 ([M+H]+, calcd. for C54H62N4O8S4Zn 1086.27), 1023.3 ([M–SO2+H]+), 894.4 ([M–3SO2]+), 830.4 ([M–4SO2]+).
Crystallographic data: C54H62N4O8S4Zn × CH2Cl2 × 0.75 C6H14, formula weight: 1238.24; temperature 233(2) K; radiation wavelength 0.71073 Å; crystal system triclinic; space group P-1 (no. 2); unit cell dimensions a = 18.5998(4) Å, b = 18.9173(3) Å, c = 21.6741(5) Å, α = 98.771(1)°, β = 104.394(1)°, γ = 109.204(1)°; volume 6743.1(2) Å3; Z = 4; density (calculated) 1.220 g/cm3; absorption coefficient 0.618 mm−1; crystal size 0.45 × 0.25 × 0.03 mm3; F(000) 2606; theta range for data collection 1.335°–24.145°; index ranges −21 ≤ h ≤ 21, −21 ≤ k ≤ 21, −24 ≤ l ≤ 24; reflections collected 36397; independent reflections 21231 [R(int) = 0.0379]; completeness to theta (24.145°) 98.5 %; absorption correction none; refinement method Full-matrix least-squares on F 2; data/restraints/parameters 21231/29/1489; goodness-of-fit on F 2 1.032; final R indices [I >2σ(I)] R 1 = 0.0769, wR 2 = 0.1998; R indices (all data) R 1 = 0.1148, wR 2 = 0.2209. Crystallographic data of 3 (Zn-(2)2) (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC no. 1453954. Copies of the data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://summary.ccdc.cam.ac.uk/structure-summary-form.
References
Falk H (1989) The chemistry of linear oligopyrroles and bile pigments. Springer Verlag, Vienna
Loudet A, Burgess K (2010) BODIPY dyes and their derivatives: syntheses and spectroscopic properties. In: Kadish KM, Smith KM, Guilard R (eds) Handbook of porphyrin science, vol 8. World Scientific Publishing, USA, p 3
Wood TE, Uddin IM, Thompson A (2010) The synthesis and properties of dipyrrins. In: Kadish KM, Smith KM, Guilard R (eds) Handbook of porphyrin science, vol 8. World Scientific Publishing, USA, p 247
Wood TE, Thompson A (2007) Chem Rev 107:1831
Filatov MA, Lebedev AY, Mukhin SN, Vinogradov SA, Cheprakov AV (2010) J Am Chem Soc 132:9552
Umezawa K, Nakamura Y, Makino H, Citterio D, Suzuki K (2008) J Am Chem Soc 130:1550
Bozdemir OA, Guliyev R, Buyukcakir O, Selcuk S, Kolemen S, Gulseren G, Nalbantoglu T, Boyaci H, Akkaya EU (2010) J Am Chem Soc 132:8029
Baudron SA (2013) Dalton Trans 42:7498
Kolemen S, Bozdemir OA, Cakmak Y, Barin G, Erten-Ela S, Marszalek M, Yum JH, Zakeeruddin SM, Nazeeruddin MK, Grätzel M, Akkaya EU (2011) Chem Sci 2:949
Li CJ, Kräutler B (2016) J Porphyrins Phthalocyanines. doi:10.1142/S1088424616500176
Li CJ, Ulrich M, Liu XJ, Wurst K, Müller T, Kräutler B (2014) Chem Sci 5:3388
Li CJ, Kräutler B (2015) Dalton Trans 44:10116
Li CJ, Fechtel M, Feng YQ, Kräutler B (2012) J Porphyr Phthalocyanines 16:556
Lightner DA (2013) Bilirubin: Jekyll and Hyde pigment of life. In: Kinghorn AD, Falk H, Kobayashi J (eds) Progress in the chemistry of organic natural products, vol 98. Springer Verlag, Vienna
Lightner DA, McDonagh AF (1984) Acc Chem Res 17:417
Moser S, Ulrich M, Müller T, Kräutler B (2008) Photochem Photobiol Sci 7:1577
Kräutler B (2014) Chem Soc Rev 43:6227
Hutchinson DW, Johnson B, Knell AJ (1973) Biochem J 133:399
Yu LH, Muthukumaran K, Sazanovich IV, Kirmaier C, Hindin E, Diers JR, Boyle PD, Bocian DF, Holten D, Lindsey JS (2003) Inorg Chem 42:6629
Hill CL, Williamson MM (1985) J Chem Soc Chem Comm 1228
Maeda H, Hasegawa M, Hashimoto T, Kakimoto T, Nishio S, Nakanishi T (2006) J Am Chem Soc 128:10024
Sakamoto R, Hoshiko K, Liu Q, Yagi T, Nagayama T, Kusaka S, Tsuchiya M, Kitagawa Y, Wong WY, Nishihara H (2015) Nat Commun 6:6713
Rieder A, Kräutler B (2000) J Am Chem Soc 122:9050
Banala S, Huber RG, Müller T, Fechtel M, Liedl KR, Kräutler B (2012) Chem Commun 48:4359
Banala S, Rühl T, Wurst K, Kräutler B (2009) Angew Chem Int Ed 48:2442
Acknowledgments
Open access funding provided by Austrian Science Fund (FWF). We thank Thomas Müller for mass spectra. The stay of C. Li in Innsbruck was funded by the China Scholarship Council. The cooperation of the two labs in Innsbruck and in Tianjin was supported by the Austrian Academic Cooperation and Mobility Unit (Project WTZ CN 14/2007) and the Tianjin International Cooperation Program of Science and Technology (Project No. 08ZCGHHZ00400). We also would like to thank the Austrian National Science Foundation (FWF, Projects I-563 and P-28522) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Prof. Ulrich Schubert on the occasion of his 70th birthday.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, C., Wurst, K., Feng, Y. et al. Synthesis, spectroscopic and crystallographic analysis of the Zn-complex of a di(β,β′-sulfoleno)pyrrin: model for Zn-complexes of bilirubin and of phylloxanthobilins. Monatsh Chem 147, 1031–1036 (2016). https://doi.org/10.1007/s00706-016-1748-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-016-1748-0