
Abstract
The clusters Ti5O(OiPr)11(OMc)(O3PR)3 (OMc = methacrylate; R = Et, CH2CH2CH2Br) and Ti10(OiPr)16(OMc)4(O3PCH2CH=CH2)10 were obtained when Ti(OiPr)4 was reacted with the corresponding bis(trimethylsilyl) phosphonate and methacrylic acid. Oxo clusters of the composition Ti6O4(OiPr)10(OMc)2(O3PR)2, with a variety of groups R (Et, Ph, CH=CH2, CH2Ph, CH2CH=CH2, CH2CH2CH2Br, CH2CH2CN, CH2C(O)Me, CH2CH2OC(O)C(Me)=CH2), were formed instead, when a stoichiometric amount of water was added to the reaction mixture.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
We have recently obtained phosphonate/acetate-substituted titanium oxo/alkoxo clusters from Ti(OiPr)4 and bis(trimethylsilyl) phosphonates in the presence of acetic acid (AcOH), which served for in situ water generation through ester formation with eliminated iPrOH. Oxo clusters of the composition Ti6O4(OiPr)10(OAc)2(O3PR)2 were obtained with a large variety of functional and non-functional substituents R (Et, CH2Ph, CH2C10H7, CH=CH2, CH2CH=CH2, CH2CH2CH2Cl, CH2CH2CH2Br), and also when the reaction conditions were varied [1]. This cluster type, which is also retained in solution, therefore appears to be very robust. Other clusters were only obtained in two exceptional cases (see below).
We extended these investigations by using methacrylic acid (McOH) instead of acetic acid. Methacrylic acid could also produce water through in situ ester formation, but would additionally provide reactive ligands in the obtained clusters and thus allow incorporating such clusters in organic polymers by polymerization with organic co-monomers (see review articles on cluster-crosslinked polymers [2, 3]). Especially the combination of ligands with different organic functionalities in one cluster appeared attractive. In this article, we report the outcome of these reactions.
Results and discussion
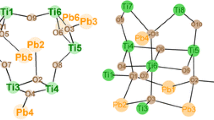
The cluster Ti5(µ3–O)(µ2–OiPr)4(OiPr)7(OMc)(O3PEt)3 (1) was formed when bis(trimethylsilyl) ethylphosphonate was reacted with methacrylic acid (McOH) and Ti(OiPr)4 in a 1:1:3 molar ratio (Fig. 1). This cluster type was previously obtained, as an exception from general outcome of the reactions with acetic acid mentioned in the “Introduction”, when bis(trimethylsilyl) 3-bromopropylphosphonate was reacted with acetic acid and Ti(OiPr)4 in a 1:1:2 ratio at room temperature. The asymmetric unit of crystalline 1 contains two independent molecules with very similar bond distances and angles.

Molecular structure of Ti5(µ3–O)(µ2–OiPr)4(OiPr)7(OMc)(O3PEt)3 (1). Hydrogen atoms are omitted for clarity. Selected bond lengths/pm and angles/°: Ti(1)–O(1) 195.98(19), Ti(1)–O(3) 196.97(19), Ti(1)–O(13) 202.08(19), Ti(1)–O(17) 178.0(2), Ti(2)–O(1) 194.0(2), Ti(2)–O(2) 195.5(2), Ti(2)–O(13) 204.4(2), Ti(3)–O(1) 196.5(2), Ti(3)–O(19) 176.8(2), Ti(4)–O(6) 218.4(2), Ti(4)–O(9) 195.4(2), Ti(5)–O(4) 195.3(2), Ti(5)–O(6) 221.2(2), Ti(5)–O(12) 208.8(2), Ti(5)–O(22) 179.8(2), P(1)–O(2) 153.8(2), P(1)–O(4) 151.5(2), P(2)–O(6) 153.9(2), P(3)–O(9) 151.8(2); Ti(2)–O(1)–Ti(1) 105.18(8), Ti(4)–O(6)–Ti(5) 98.03(7)
The structure of 1 is related to that of the clusters Ti4(µ3–O)(µ2–OiPr)3(OiPr)5(O3PR)3L (L = neutral ligand) [4–7], which consist of a symmetrical Ti3(μ3–O)(μ2-OiPr)3(OiPr)3 unit (Ti(1)–Ti(3) in Fig. 1 to which a Ti(OiPr)2L group is connected by means of three phosphonate ligands. In 1, the capping Ti(OiPr)2L group is replaced by a Ti2(µ2–OiPr)(OiPr)4(µ2–OMc) moiety (Ti(4) and Ti(5) in Fig. 1). Two of the phosphonate ligands are coordinated to only one Ti atom of the Ti2 unit and have a 3.111 binding mode (w.xyz refers to the number of metal atoms to which the phosphonate ligand is coordinated [w], and the number of metal atoms to which each oxygen is coordinated [x, y, z] [8]), while the third bridges both of them and has a binding mode of 4.211. The degree of condensation of 1 is 0.2 (O/Ti ratio of the cluster core), while it is 0.67 for the clusters Ti6O4(OiPr)10(OAc)2(O3PR)2 obtained with acetic acid under the same conditions. This indicates that ester + water formation of methacrylic acid, relative to the rate of substitution [9], is slower than that of acetic acid.
1H, 13C, and 31P NMR spectra of re-dissolved crystals of 1 in C6D6 showed numerous signals. In the 31P NMR spectrum, for example, eight resonances were observed, while two signals are expected if the solid-state structure of 1 was retained in solution. We therefore assume that 1 is in equilibrium with other compounds.
Isostructural Ti5(µ3–O)(µ2–OiPr)4(OiPr)7(OMc)(O3PCH2CH2CH2Br)3 (2) was obtained from the reaction of bis(trimethylsilyl) bromopropylphosphonate, methacrylic acid, and Ti(OiPr)4 in a ratio of 1:2:3. The higher proportion of McOH thus did not influence the outcome of the reaction. Ti2(OMc)2(OiPr)6 iPrOH [8] was formed as a by-product, as proven by single crystal XRD. The 1H and 31P NMR spectra of the solid residue correspondingly showed numerous signals. Therefore, it can be assumed that a mixture of products was obtained and/or several species are in equilibrium with each other.
When a 1:2:3 mixture of bis(trimethylsilyl) allylphosphonate, methacrylic acid, and Ti(OiPr)4 was heated to reflux, the complex Ti10(µ2–OiPr)2(OiPr)14(OMc)4(O3PCH2CH=CH2)10 (3) (Fig. 2) was obtained after crystallization from CH2Cl2. It is noteworthy that 3 contains no oxo groups, but more OiPr groups were substituted by OMc or O3PR ligands compared to 1 and 2. The different outcome of this reaction, compared to 1 and 2, may be due to the higher reaction temperature. We have previously shown that higher reaction temperatures favor substitution over ester formation [9].

Molecular structure of Ti10(µ2–OiPr)2(OiPr)14(OMc)4(O3PCH2CH = CH2)10 (3). Hydrogen atoms are omitted for clarity. Selected bond lengths/pm and angles/°: Ti(1)–O(2) 193.6(2), Ti(1)–O(3) 195.9(2), Ti(1)–O(4) 202.1(2), Ti(1)–O(8) 195.8(2), Ti(1)–O(12) 200.9(2), Ti(1)–O(20) 175.6(2), Ti(2)–O(7) 216.5(2), Ti(2)–O(19) 206.3(3), Ti(3)–O(9) 193.5(2), Ti(3)–O(10) 221.1(2), Ti(4)–O(5) 193.1(2), Ti(5)–O(7) 217.4(2), Ti(5)–O(18) 206.8(3); Ti(2)–O(7)–Ti(5) 99.38(9), Ti(2)–O(21)–Ti(5) 110.3(1), Ti(3)–O(10)–Ti(4) 126.8(1), O(1)–P(1)–O(2) 110.7(1), O(1)–P(1)–C(1A) 107.3(2), O(10)–P(4)–O(11) 99.1(1), O(10)–P(4)–O(12) 114.6(1), O(11)–P(4)–O(12) 115.6(1)
The structure of 3 consists of two Ti5(OiPr)8(OMc)2(O3P-allyl)5 units, which are bridged by two (3.111) phosphonate ligands. The Ti5 units are composed of methacrylate-bridged dimers Ti2(µ2–OiPr)(OiPr)3(OMc) (Ti(2), Ti(5)) and Ti2(OiPr)3(OMc) (Ti(3), Ti(4)), respectively, which are connected through phosphonate ligands among each other as well as to the fifth titanium atom (Ti(1)). Each of the octahedrally coordinated titanium atoms is at least bound to two different phosphonate ligands; Ti(1) is coordinated by five different oxygen atoms of phosphonate ligands and one OiPr ligand. The complexity of the structure of 3 is also reflected in the different binding modes of the phosphonate ligands, of which six are 3.111, two are 3.211, and two are 4.211.
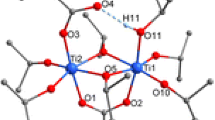
The reactions leading to 1, 2, and 3 show that clusters with a noticeably lower degree of condensation were formed compared to analogous reactions with acetic acid [1]. This is most probably due to the lower reaction rate of ester formation between methacrylic acid and isopropyl alcohol compared to that of acetic acid [10]. This assumption was proven by the deliberate addition of water to the reaction mixture. Thus, when bis(trimethylsilyl) 3-bromopropylphosphonate, methacrylic acid, Ti(OiPr)4, and water were reacted in a 1:1:3:2 ratio, the cluster Ti6O4(OiPr)10(OMc)2(O3PCH2CH2CH2Br)2 (4) (Fig. 3) was obtained. The cluster 4 is isostructural to Ti6O4(OiPr)10(OAc)2(O3PCH2CH2CH2Br)2 obtained with acetic acid [1].

Molecular structure of Ti6O4(OiPr)10(OMc)2(O3PCH2CH2CH2Br)2 (4). Hydrogen atoms are omitted for clarity. Selected bond lengths/pm and angles/°: Ti(1)–O(1) 197.1(1), Ti(1)–O(3) 208.8(1), Ti(1)–O(5) 199.0(1), Ti(1)–O(6) 196.1(1), Ti(1)–O(7) 177.7(1), Ti(1)–O(12) 192.9(1), Ti(2)–O(1) 199.2(1), Ti(2)–O(2) 187.3(1), Ti(2)–O(10) 196.5(1), Ti(3)–O(1) 189.9(1), Ti(3)–O(2)* 175.0(1), Ti(3)–O(6) 204.7(1), Ti(3)–O(9) 180.9(1); Ti(3)–O(1)–Ti(1) 105.83(6), Ti(3)–O(1)–Ti(2) 149.36(7), Ti(1)–O(1)–Ti(2) 104.15(6), Ti(3)–O(2)–Ti(2) 148.49(8), Ti(1)–O(5)–Ti(2) 102.48(6), Ti(1)–O(6)–Ti(3) 100.75(5)
The centrosymmetric cluster 4 is isostructural to the previously reported acetate-substituted clusters Ti6O4(OiPr)10(OAc)2(O3PR)2 [1]. The cluster core is formed by two parallel, unsymmetrically substituted Ti3(µ3–O)(µ2–OiPr)2(OiPr)3(µ2–OMc) units connected by µ2-oxo (O(2) and O(2)*, * denotes symmetry-related atoms) and phosphonate bridges. Ti(1) and Ti(2) are bridged by both an OiPr and a methacrylate ligand and are octahedrally coordinated while Ti(3) has a distorted trigonal bipyramidal coordination sphere. The central Ti3O unit is unsymmetrical, with one short (Ti(3)–O(1) 189.9(1) pm) and two long Ti–O distances (Ti(1)–O(1) 197.1(1), Ti(2)–O(1) 199.2(2) pm), as in the acetate derivatives Ti6O4(OiPr)10(OAc)2(O3PR)2.
NMR data show that the structure of 4, especially also their inversion symmetry is retained in solution. Thus, one signal was observed in the 31P NMR spectrum at 27.34 ppm. In the 1H NMR spectrum five doublets for the methyl groups of the OiPr ligands were observed and three signals for the CH groups (at 4.86, 4.97, and 5.33 ppm) the latter two with double intensity. One singlet at 2.08 ppm and two multiplets at 5.41 and 6.36 ppm can be assigned to the two OMc ligands. In the 13C NMR spectrum only one doublet for each P-CH2 group was found and one set of signals for the OMc ligands. The signals of the OiPr ligands were partly overlapping.
The clusters 5–12 with a great variety of functional or non-functional phosphonate ligands were obtained according to Scheme 1 by the same synthesis procedure as that for 4. The 1H, 31P, and 13C NMR spectra of 5–12 are similar to that of 4.

Conclusions
The first step in reactions of metal alkoxides with carboxylic or phosphonic acids is the substitution of an OR ligand by a carboxylate or phosphonate ligand. The thus liberated alcohol can undergo ester formation with the carboxylic or phosphonic acid, which produces water that hydrolyzes part or all of the remaining M–OR groups. Thus two reactions, viz. substitution and ester formation, compete with each other, and their relative rate is one of the decisive parameters influencing the outcome of such reactions. How the clusters are formed from the initially formed M(OR) x (carboxylate/phosphonate) y derivatives has not been elucidated in any case. The situation becomes even more complex when two different metal alkoxides or, as in the present case, two different acids are involved.
In previous work, we had preferentially obtained phosphonate/acetate-substituted titanium oxo/alkoxo clusters of the composition Ti6O4(OiPr)10(OAc)2(O3PR)2 from Ti(OiPr)4 and bis(trimethylsilyl) phosphonates in the presence of acetic acid (AcOH) [1]. The results of the work reported in this article show that the degree of condensation of the obtained clusters (1 and 2) was lower when acetic acid was replaced by methacrylic acid. In one case, the product (compound 3) contained no oxo groups at all. This can be taken as evidence that the rate of esterification of methacrylic acid is lower than that of acetic acid.
The lower esterification rate can be compensated, however, by controlled addition of a stoichiometric amount of “external” water. The thus obtained methacrylate/phosphonate-substituted clusters 4–12, with a very wide variety of phosphonate ligands, are isostructural to the acetate-substituted clusters Ti6O4(OiPr)10(OAc)2(O3PR)2 obtained in earlier experiments [1]. Incorporation of the polymerizable OMc ligands is a very interesting option for the preparation of cluster-crosslinked polymers [2, 3], especially because this allows the combination (a) of reactive and non-reactive ligands as well as (b) ligands with different organic functionalities in a controlled manner in one cluster.
Experimental
All operations were carried out in a moisture- and oxygen-free argon atmosphere using Schlenk techniques. Isopropyl alcohol was dried by refluxing twice over sodium metal and distillation. The bis(trimethylsilyl) phosphonates were prepared as reported before [1].
Methacrylate-phoshonate-substituted Ti5 oxo clusters
Ti5O(OiPr)11(OMc)(O3PEt)3 (1): 1.6 cm3 of Ti(OiPr)4 (5.42 mmol) was added to a solution of 500 mm3 of bis(trimethylsilyl) ethylphosphonate (1.81 mmol) and 153 mm3 of methacrylic acid (1.81 mmol) in 2 cm3 of isopropyl alcohol. Crystals of 1 were obtained from this solution after 8 weeks. Yield 410 mg (35 %).
Ti5O(OiPr)11(OMc)(O3PCH2CH2CH2Br)3 (2): 1.2 cm3 of Ti(OiPr)4 (4.1 mmol) was added to a solution of 400 mm3 of bis(trimethylsilyl) 3-bromopropylphosphonate (1.34 mmol) in 2 cm3 of iPrOH, followed by addition of 113 mm3 of methacrylic acid (1.34 mmol). Crystals of 2 were obtained from this solution after 3 weeks. Yield 620 mg (mixture of compounds).
Methacrylate-phoshonate-substituted Ti10 oxo cluster Ti10(OiPr)16(OMc)4(O3PCH2CHCH2)10 (3):
7.1 cm3 of Ti(OiPr)4 (24 mmol) was added to a solution of 2 cm3 of bis(trimethylsilyl) allylphosphonate (8 mmol) and 1.35 cm3 of methacrylic acid (16 mmol) in 12 cm3 of isopropyl alcohol. The solution was heated to reflux for 16 h, and a suspension was formed. The solid was separated by filtration and recrystallized from CH2Cl2. Yield 120 mg (5 %). The crystals could not be re-dissolved in CD2Cl2 or another non-coordinating organic solvent and therefore no NMR measurements were performed.
Methacrylate-phoshonate-substituted Ti6 oxo clusters: Ti6O4(OiPr)10(OMc)2(O3PCH2CH2CH2Br)2 (4) and Ti6O4(OiPr)10(OMc)2(O3PPh)2 (5)
Ti(OiPr)4 (1.17 cm3, 4 mmol) was quickly added to a solution of 400 mm3 of bis(trimethylsilyl) 3-bromopropylphosphonate (1.3 mmol) [or 225 mg of bis(trimethylsilyl) phenylphosphonate (0.82 mmol)] in 2 cm3 of 2-propanol followed by addition of 110 mm3 of methacrylic acid (1.3 mmol). Finally, 48 mm3 of water (2.7 mmol) diluted in 1 cm3 of 2-propanol was injected quickly directly into the solution. Crystals were obtained after 1 week.
4: yield 420 mg (43 %); 1H NMR (C6D6, 250 MHz): δ = 1.31 (d, 3 J H,H = 6.09 Hz, 12H, CHMe), 1.41 (d, 3 J H,H = 6.24 Hz, 12H, CHMe), 1.48 (d, 3 J H,H = 6.09 Hz, 12H, CHMe), 1.74 (d, 3 J H,H = 6.24 Hz, 12H, CHMe), 1.82 (d, 3 J H,H = 6.24 Hz, 12H, CHMe), 1.72–1.88 (m, 2H, PCH2), 2.08 (s, 6H, =CCH3), 2.36 (m, 3 J P,H = 15.84 Hz, 3 J H,H = 7.31 Hz, 2H, CH 2 CH2P), 3.46 (t, 3 J H,H = 7.16 Hz, CH2Br), 4.86 (m, 3 J H,H = 6.17 Hz, 2H, OCH), 4.97 (m, 3 J H,H = 6.13 Hz, 4H, OCH), 5.33 (m, 3 J H,H = 6.20 Hz, 4H, OCH), 5.39–5.43 (m, 2H, =CH2), 6.34–6.38 (m, 2H, =CH2) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 27.34 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.63 (CHMe), 23.89 (CHMe), 24.20 (CHMe), 24.81 (CHMe), 25.20 (CHMe), 25.67 (d, 1 J P,C = 157 Hz, PCH2), 27.68 (d, 2 J P,C = 4.53 Hz, CH 2 CH2P), 33.81 (d, 3 J P,C = 14.96 Hz, CH2Br), 77.83 (OCH), 78.69 (OCH), 79.40 (OCH), 123.41 (=CH2), 140.02 (=CMe–), 173.39 (COO) ppm.
5: yield 160 mg (27 %); 1H NMR (C6D6, 250 MHz): δ = 1.26 (d, 3 J H,H = 6.13 Hz, 12H, CHMe), 1.43 (d, 3 J H,H = 6.08 Hz, 24H, CHMe), 1.82 (d, 3 J H,H = 6.40 Hz, 12H, CHMe), 1.84 (d, 3 J H,H = 6.40 Hz, 12H, CHMe), 2.14 (s, 6H, CH3 (OMc)), 4.88 (m, 3 J H,H = 6.17 Hz, 2H, CH (OiPr)), 4.99 (m, 3 J H,H = 6.13 Hz, 4H, CH (OiPr)), 5.36–5.58 (m, 6H, CH (OiPr) + CH2 (OMc)), 6.43–6.46 (m, 2H, CH2 (OMc)), 7.10–7.28 (m, 2H, CH (Ph)), 7.34–7.44 (m, 4H, Ph), 8.33–8.44 (m, 4H, Ph) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 16.03 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.64 (CHMe), 24.06 (CHMe), 24.26 (CHMe), 24.83 (CHMe), 25.17 (CHMe), 77.93 (OCH), 78.78 (OCH), 79.48 (OCH), 123.28 (=CH2), 130.25 (d, J P,C = 2.99 Hz, Ph), 130.99 (d, J P,C = 9.36 Hz, Ph), 131.80 (d, J P,C = 9.97 Hz, Ph), 134.50 (d, 1 J P,C = 208.43 Hz, Ph), 140.25 (=CMe–), 173.46 (COO) ppm.
Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 P–CH=CH 2 ) 2 ( 6 ), Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 PCH 2 Ph) 2 ( 7 ), Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 PCH 2 –CH=CH 2 ) 2 ( 8 ), Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 PCH 2 CH 2 –OMc) 2 ( 9 ), Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 PCH 2 CH 2 C≡N) 2 ( 10 ), Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 PCH 2 CH 3 ) 2 ( 11 ), Ti 6 O 4 (OiPr) 10 (OMc) 2 (O 3 PCH 2 COCH 3 ) 2 ( 12 ).
Compared to 4 and 5, the synthesis was slightly modified. In the synthesis of 6 660 mm3 of Ti(OiPr)4 (2.27 mmol) was added to a mixture of 200 mm3 of bis(trimethyl)silyl vinylphosphonate (0.76 mmol) and 64 mm3 of methacrylic acid (0.76 mmol) in 2 cm3 of 2-propanol. Immediately afterwards, 27.3 mm3 of water (1.52 mmol) diluted in 0.5 cm3 of 2-propanol was added. Crystals of 6 were obtained after 3 days. The syntheses of 7–11 were analogous. The synthesis of 12 was done analogously, but the precursor solution was additionally heated after addition of water until a clear solution was obtained.
6: yield 70 mg (14 %); 1H NMR (C6D6, 250 MHz): δ = 1.33 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.41 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.51 (d, 3 J H,H = 5.94 Hz, 12H, OCHMe), 1.79 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.84 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 2.09 (s, 6H, =CMe), 4.87 (m, 3 J H,H = 6.13 Hz, 2H, OCH), 5.02 (m, 3 J H,H = 5.90 Hz, 4H, OCH), 5.30–5.45 (m, 6H, OCH + =CH2), 5.74 (ddd, 2 J H,H = 3.50 Hz, 3 J H,H = 12.03 Hz, 3 J P,H = 49.42 Hz, 4H, OCH), 6.21–6.57 (m, 6H, CH2(vinyl) + CH(vinyl) + =CH2 (OMc)) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 14.25 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.58 (=CMe), 23.93 (OCHMe), 24.23 (OCHMe), 24.78 (OCHMe), 25.17 (OCHMe), 77.82 (OCH), 78.65 (OCH), 79.38 (OCH), 123.16 (=CH2 (OMc), 128.66 (=CH2 (vinyl)), 130.81 (d (1 J P,C = 204.2 Hz), CH (vinyl)), 140.22 (=CMe), 173.41 (COO) ppm.
7: yield 160 mg (34 %); 1H NMR (C6D6, 250 MHz): δ = 1.30 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.41–1.49 (m, 24H, OCHMe), 1.65 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.72 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 2.07 (s, 6H, = CMe), 3.20 (d, 2 J P,H = 22.69 Hz, 4H, PCH2), 4.83–5.02 (m, 3 J H,H = 6.07 Hz, 6H, OCH), 5.18–5.33 (m, 3 J H,H = 6.24 Hz, 6H, OCH), 5.40 (br, 2H, = CH2), 6.32 (br, 2H, = CH2), 7.18–7.28 (m, 2H, Ph), 7.31–7.39 (m, 4H, Ph), 7.63–7.68 (m, 4H, Ph) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 23.34 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.64 (=CMe), 23.81 (OCHMe), 24.15 (OCHMe), 24.95 (OCHMe), 25.26 (OCHMe), 35.18 (d, 1 J P,C = 152.25 Hz, PCH2), 77.74 (OCH), 78.69 (OCH), 79.05 (OCH), 123.12 (CH2 (OMc), 125.83 (d, J P,C = 3.00 Hz, Ph), 130.44 (d, J P,C = 6.98 Hz, Ph), 134.88 (d, J P,C = 8.98 Hz, Ph), 140.19 (=CMe–), 173.34 (COO) ppm.
8: yield 180 mg (33 %); 1H NMR (C6D6, 250 MHz): δ = 1.31 (d, 3 J H,H = 6.10 Hz, 12H, OCHMe), 1.41 (d, 3 J H,H = 6.10 Hz, 12H, OCHMe), 1.50 (d, 3 J H,H = 6.03 Hz, 12H, OCHMe), 1.77 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.84 (d, 3 J H,H = 6.21 Hz, 12H, OCHMe), 2.09 (s, 6H, =CMe), 2.68 (dd, 2 J P,H = 22.69 Hz, 3 J H,H = 7.08 Hz, 4H, PCH2), 4.85 (m, 3 J H,H = 6.09 Hz, 2H, OCH), 5.00 (m, 3 J H,H = 6.05 Hz, 4H, OCH), 5.22–5.45 (m, 10H, OCH + CH2 (OMc) + CH2 (allyl)), 6.16–6.35 (m, 2H, CH (allyl)), 6.38 (br, 2H, = CH2 (OMc)) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 24.16 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.59 (=CMe), 23.95 (OCHMe), 24.21 (OCHMe), 24.75 (OCHMe), 25.17 (OCHMe), 33.31 (d, 1 J P,C = 154.33 Hz, PCH2), 77.70 (OCH), 78.59 (OCH), 79.31 (OCH), 117.29 (d, 3 J P,C = 14.96 Hz, = CH2), 123.14 (CH2 (OMc)), 130.96 (d, 2 J P,C = 10.97 Hz, =CH), 140.20 (C (OMc)), 173.28 (COO) ppm.
9: yield 100 mg (22 %); 1H NMR (C6D6, 250 MHz): δ = 1.31 (d, 3 J H,H = 6.10 Hz, 12H, OCHMe), 1.42 (d, 3 J H,H = 6.15 Hz, 12H, OCHMe), 1.50 (d, 3 J H,H = 6.08 Hz, 12H, OCHMe), 1.76 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.82 (d, 3 J H,H = 6.20 Hz, 12H, OCHMe), 1.89 (s, 6H, CH3 (OMc ester)), 2.10 (s, 6H, =CMe), 2.33–2.50 (m, 4H, PCH2), 4.79–5.06 (m, 10H, OCH + CH2O), 5.26 (br, 2H, CH2 (OMc ester)), 5.36 (m, 3 J H,H = 6.24 Hz, 4H, OCH), 5.43 (br, 2H, =CH2), 6.20 (br, 2H, CH2 (OMc ester)), 6.38 (br, 2H, =CH2) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 23.59 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.03 (CH3 (OMc ester)), 18.60 (=CMe), 23.90 (OCHMe), 24.19 (OCHMe), 24.71 (OCHMe), 25.16 (OCHMe), 27.88 (d, 1 J P,C = 152.46 Hz, PCH2), 60.92 (d, 2 J P,C = 3.98 Hz, OCH2), 78.03 (OCH), 78.80 (OCH), 79.71 (OCH), 123.67 (CH2 (OMc), 124.64 (CH2 (OMc ester)), 136.68 (C (OMc ester)), 139.94 (=CMe–), 166.56 (COO (OMc ester)), 173.50 (COO) ppm.
10: yield 120 mg (24 %); 1H NMR (C6D6, 250 MHz): δ = 1.26 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.38 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.41 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.67 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.77 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.80–1.89 (m, 4H, CH2CN), 2.02 (s, 6H, =CMe), 2.54–2.66 (m, 4H, PCH2), 4.79 (m, 2H, OCH), 4.89 (m, 4H, OCH), 5.29 (m, 4H, OCH), 5.36 (br, 2H, =CH2), 6.29 (br, 2H, = CH2) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 24.16 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 11.67 (d, 2 J P,C = 2.50 Hz, CH 2 CN), 18.46 (=CMe), 22.04 (PCH2), 23.80 (OCHMe), 24.16 (OCHMe), 24.63 (OCHMe), 25.05 (OCHMe), 25.11 (OCHMe), 78.13 (OCH), 78.94 (OCH), 79.96 (OCH), 118.86 (d, 3 J P,C = 18.95 Hz, CN), 123.61 (=CH2), 139.83 (=CMe–), 173.48 (COO) ppm.
11: yield 230 mg (48 %); 1H NMR (C6D6, 250 MHz): δ = 1.23–1.47 (m, 6H, CH 3 CH2P), 1.32 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.41 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.50 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.65–1.93 (m, 4H, PCH2), 1.77 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.84 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 2.09 (s, 6H, =CMe), 4.86 (m, 3 J H,H = 6.09 Hz, 2H, OCH), 5.00 (m, 3 J H,H = 6.09 Hz, 4H, OCH), 5.27–5.37 (m, 4H, OCH), 5.39 (br, 2H, CH2 (OMc)), 6.36 (br, 2H, CH2 (OMc)) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 29.82 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 7.42 (d, 2 J P,C = 6.28 Hz, CH 3 CH2P), 18.58 (=CMe), 20.00 (d, 1 J P,C = 158.57 Hz, PCH2), 23.93 (OCHMe), 24.20 (OCHMe), 24.75 (OCHMe), 25.13 (OCHMe), 25.21 (OCHMe), 77.51 (OCH), 78.44 (OCH), 79.09 (OCH), 122.99 (=CH2), 140.28 (=CMe–), 173.23 (COO) ppm.
12: yield 260 mg (53 %); 1H NMR (C6D6, 250 MHz): δ = 1.32 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.39 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.48 (d, 3 J H,H = 6.09 Hz, 12H, OCHMe), 1.71 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 1.80 (d, 3 J H,H = 6.24 Hz, 12H, OCHMe), 2.05 (s, 6H, =CMe), 2.46 (m, 4H, MeCO), 3.00 (d, 2 J P,H = 23.91 Hz, 4H, PCH2), 4.81 (m, 3 J H,H = 6.17 Hz, 2H, OCH), 4.98 (m, 3 J H,H = 6.13 Hz, 4H, OCH), 5.31 (m, 3 J H,H = 6.32 Hz, 4H, OCH), 5.38 (br, 2H, CH2 (OMc)), 6.32 (br, 2H, =CH2) ppm; 31P NMR (C6D6, 101.2 MHz): δ = 18.69 ppm; 13C NMR (C6D6, 62.9 MHz): δ = 18.51 (=CMe), 23.80 (OCHMe), 24.19 (OCHMe), 24.72 (OCHMe), 25.20 (OCHMe), 30.50 (CH 3 –CO), 45.25 (d, 1 J P,C = 139.17 Hz, PCH2), 78.29 (OCH), 78.94 (OCH), 79.90 (OCH), 123.59 (CH2 (OMc)), 139.89 (C (OMc)), 173.48 (COO), 199.11 (d, 2 J P,C = 5.58 Hz, C=O) ppm.
X-ray structure analyses
All measurements were performed using MoK α radiation (λ = 71.073 pm). Data were collected on a Bruker Axs Smart Apex II four-circle diffractometer with κ-geometry at 100 K with φ and ω-scans and 0.5° frame width (Table 1) and corrected for polarization and Lorentz effects. An empirical absorption correction (Sadabs) was applied. The cell dimensions were refined with all unique reflections. Saint Plus (Bruker Analytical X-ray Instruments, 2007) was used to integrate the frames. Symmetry was checked with the program Platon.
The structures were solved by the Patterson method (Shelxs97). Refinement was performed by the full-matrix least-squares method based on F 2 (Shelxl97) with anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were inserted in calculated positions and refined riding with the corresponding atom. In 1, 3, 4, 6–9, 11, and 12 OiPr ligands were disordered. In 6 and 12 one OiPr ligand was additionally refined for three different positions. In 6, 9, and 11 the methacrylate ligand was bridging either between Ti(1) and Ti(2) or between Ti(1) and Ti(3). Two allyl groups in 3 and one Br atom in 2 were also disordered.
CCDC-1027711 (for 1), -1027712 (for 2), -1027713 (for 3), -1027714 (for 4), -1027715 (for 5), -1027716 (for 6), -1027717 (for 7), -1027718 (for 8), -1027719 (for 9), -1027720 (for 10), -1027721 (for 11), and -1027722 (for 12) contain the supplementary crystallographic data. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
References
Czakler M, Artner C, Schubert U (2014) Eur J Inorg Chem 2038
Schubert U (2001) Chem Mater 13:3487
Schubert U (2011) Chem Soc Rev 40:575
Guerrero G, Mehring M, Mutin PH, Dahan F, Vioux A (1999) J Chem Soc Dalton Trans 61:1537
Mehring M, Guerrero G, Dahan F, Mutin PH, Vioux A (2000) Inorg Chem 39:3325
Chakraborty D, Chandrasekhar V, Bhattacharjee M, Krätzner R, Roesky HW, Noltemeyer M, Schmidt H (2000) Inorg Chem 39:23
Czakler M, Artner C, Schubert U (2013) Eur J Inorg Chem 5790
Chandrasekhar V, Senapati T, Dey A, Hossain S (2011) Dalton Trans 40:5394
Czakler M, Artner C, Schubert U (2014) Monatsh Chem. doi:10.1007/s00706-015-1443-6
Newman MS (1950) J Am Chem Soc 72:4783
Acknowledgments
This work was supported by the Fonds zur Förderung der wissenschaftlichen Forschung (FWF), Austria (Project P22915). The X-ray measurements were carried out at the X-ray Center of Vienna University of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Czakler, M., Artner, C. & Schubert, U. Titanium oxo/alkoxo clusters with both phosphonate and methacrylate ligands. Monatsh Chem 146, 1249–1256 (2015). https://doi.org/10.1007/s00706-015-1444-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1444-5