
Abstract
A series of novel phenylsulfonyl- and 4-aminophenylsulfonyl-carboximidamides were synthesized by condensation of sulfonamides with heterocyclic methyl carbimidates obtained from heterocyclic carbonitriles and used ‘at its inception.’ The molecular structure of the obtained compounds is discussed. Compounds possessing heterocyclic systems with a nitrogen atom in the α position to the functional group showed a different single-crystal structure than expected. The synthesized derivatives were evaluated for antimicrobial activities: tuberculostatic, antibacterial, and antifungal.
Graphical Abstract

.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
At the end of the twentieth century, a number of new and “reemerging” pathogens were recognized [1]. These included S. pneumonia, L. pneumophila, M. avium, E. coli, H. pylori, S. aureus, C. albicans, and M. tuberculosis [2–4]. These microorganisms quickly develop a multidrug resistance (MDR) to used chemotherapeutics and antibiotics. A special case is M. tuberculosis, whose strains also develop extensive drug-resistance (XDR). Resistant strains of microorganisms are a major threat to immunocompromised individuals, and infections caused by them are the most common complication in HIV-positive patients [5]. At the same time, a lack of development of new antimicrobial drugs is observed, which can pose a serious threat to public health [6]. Thus, the interest of many research groups is focused on the search for new drugs active against resistant strains.
One of the research directions is to modify the structure of already used drugs. So the interest in chemical groups such as, for example, sulfonamides has not diminished. This group is characterized by multidirectional pharmacological activity. Sulfonamides act as anhydrase inhibitors [7], antifungal [8], antiviral [9], anticancer [10], anti-inflammatory [11], and of course antibacterial agents.
Multidirectional biological activity also characterizes compounds possessing an amidine functional group. Amidine derivatives have anti-degenerative [12], antitumor [13], and anti-platelet effects [14]. Compounds with anti-HIV, antibacterial, and antifungal activities have also been found among them [15, 16].
There are few reports on the pharmacological activity of sulfonamidines. So far, only their in vitro ability to compete with triiodothyronine for binding to the thyroid hormone-α1 receptor (hTHR-α1) has been described [17]. These compounds can be obtained in several ways. They are formed as a result of the reaction of carbonitriles with primary sulfonamides [18] or in a reaction of amidines with sulfonyl chlorides [19]. The reports of reactions of sulfonamides with alkyl- or phenylcarbimidates could also be found in the chemical literature [20]. In the structure assigned to the products, two protons are connected to different nitrogen atoms of the amidine moiety [21]. That structure was adopted on the basis of 1H NMR spectra in which two different signals for those protons were observed. The reaction of sulfonamides with heterocyclic carbimidates has not been described so far.
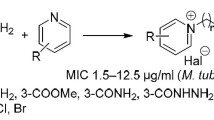
The above facts prompt us to synthesize sulfonyl-carboximidamides possessing in their structure phenylsulfonyl or 4-aminophenylsulfonyl moieties linked to heterocyclic rings of pyridine, pyrimidine, or pyrazine by the sulfonamidine group. Synthesized compounds have been evaluated for their antimicrobial activity in vitro: tuberculostatic, antibacterial, and antifungal.
Results and discussion
The subject of this work was the synthesis of heterocyclic phenylsulfonyl- and 4-aminophenylsulfonyl-carboximidamides 1–13. The performed reactions are shown in Scheme 1.

The presented method of synthesis uses an intermediate such as carbimidate “at its inception,” and this is its main advantage. Carbimidates were obtained from the corresponding carbonitriles in methanol in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and without isolation underwent further reaction with benzene sulfonamide or 4-aminobenzenesulfonamide. The isolated carbimidates were used for the synthesis of pyrazine (10, 11) and 6-methoxypyrazine (12, 13) derivatives. They were obtained easily from 2-cyanopyrazine and 6-chloro-2-cyanopyrazine, respectively [22, 23]. Carbimidates were refluxed with benzene sulfonamide or 4-aminobenzenesulfonamide in diglyme (2-methoxyethyl ether) solution. All reactions proceeded with yields from moderate (38 %) to very good (83 %).
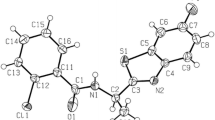
The structures of all these new compounds were confirmed by IR and NMR spectra as well as elemental analyses. Two signals for the NH groups shifted from each other have been observed in the 1H NMR spectra. These separated signals can be due to the taken amino-imine structure of compounds obtained (Fig. 1, structure a) as we suggest for 3- and 4-pyridine derivatives 3–6. They can also be a result of the magnetic inequivalence of NH protons in the amine moiety upon formation of a hydrogen bond in the case of the heterocyclic compounds in which the amidine group is in the α position to the nitrogen atom of heterocyclic ring (structure b). X-ray diffraction analysis was performed for N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide to address that question. We have described the synthesis of this compound previously [24]. It was chosen because we were able to obtain its crystals of sufficient size. The results of the single-crystal diffraction study confirmed a tautomeric structure b (Fig. 1). If both hydrogen atoms are bonded to the same nitrogen atom in the solid state, their magnetic inequivalence in the solution is probably caused by formation of hydrogen bonds and reduction of symmetry.

Possible systems of intramolecular hydrogen bonds in target molecules
The study also resolved the molecular structure of the products of the reaction between carbimidates and 4-aminobenzenesulfonamide, as the active group in that reaction could be both the amine group of the sulfonamide moiety, as it was in the case of the reaction of benzene sulfonamide, or the aromatic amine group in the para position to the sulfonamide moiety, since the reactions of aromatic and aliphatic amines with carbimidates have been described [25, 26]. For that purpose, the reaction of methyl pyrazine-2-carbimidate with N-(4-sulfamoylphenyl)acetamide was carried out. The resulting product 9 was identical with the compound that was obtained by the acetic anhydride acylation of derivative 8, which was formed in the reaction of methyl pyrazine-2-carbimidate with 4-aminobenzenesulfonamide. This showed that the sulfonamide group was the active group in the reactions carried out, and the resulting compounds had structure c (Fig. 2).

Structure of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide showing 25 % probability displacements for ellipsoids. H atoms are shown as small spheres of arbitrary radius (intramolecular N–H···O and N–H···N interactions are represented by dashed lines)
Crystal structure of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide
The crystallographic data, data collection, and structure refinement of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide are summarized in Table 1. The bond lengths and angles characterizing the geometry of the molecules are presented in Table 2.
N′-(4-Aminophenylsulfonyl)-4-chloropicolinimidamide crystallized in the P21/c monoclinic space group, with a = 14.6885(7) Å, b = 5.7930(3) Å, c = 16.0421(9) Å, and β = 97.530(5)°, Z = 4, and V = 1353.25(12) Å3.
In the molecule of the title compound (Fig. 2), the bond lengths and angles characterizing the geometry of the 4-aminophenylsulfonyl and pyridine fragments are typical for compounds containing them (Table 2).
In the crystal structure of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide, the H atoms from the amino group bonded with the C7 atom participate in the intramolecular N16–H16A···O18 and N16–H16B···N1 hydrogen bond (Table 3; Fig. 2). In the packing, the molecules are linked into chains of rings along the c axis (Fig. 3b). In these rings, four molecules of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide are linked via N19–H19A···O17 and N19–H19B···N19 and form the \( R_{4}^{4} \) (20) hydrogen bond ring motif (Fig. 3a). The parallel lying chains of rings are connected through the N16–H16A···O17 hydrogen bond and form columns along the b axis (Table 3; Fig. 3a). In the crystal lattice, these columns form a zipper-type supramolecular motif.

The \( R_{4}^{4} \) (20) hydrogen bond motif (a) and the arrangement of the molecules in the crystal structure of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide viewed along b axis (b). Dashed lines: N–H···O and N–H···N interactions. H atoms not involved in interactions omitted
Tuberculostatic activity
The synthesized phenylsulfonyl- and 4-aminophenylsulfonyl-carboximidamides 1–13 were examined in vitro for their tuberculostatic activity against M. tuberculosis H37Rv strain and two “wild” strains isolated from tuberculosis patients: one (Spec. 210) resistant to p-aminosalicylic acid (PAS), isonicotinic acid hydrazide (INH), etambutol (ETB), and rifampicine (RFP), and the other (Spec. 192) fully sensitive to the administered tuberculostatics (Table 4).
Investigations were performed by a classical test-tube method of successive dilution in Youmans’ modification of Proskauer and Beck’s liquid medium containing 10 % of bovine serum [33, 34]. Bacterial suspensions were prepared from 14-day-old cultures of slowly growing strains and from 48-h-old cultures of saprophytic strains [35, 36]. Solutions of the compounds in ethylene glycol were tested. Stock solutions contained 10 mg of compounds in 1 cm3. Dilutions (in geometric progression) were prepared in Youmans’ medium. The medium containing no investigated substances and containing isoniazid (INH) as a reference drug were used for comparison. Incubation was performed at a temperature of 37 °C. The MIC values were determined as minimum concentration inhibiting the growth of tested tuberculous strains in relation to the probe with no tested compound. The influence of the compound on the growth of bacteria at a certain concentration, 3.1, 6.2, 12.5, 25, 50, and 100 μg/cm3, was evaluated.
The study showed that the newly synthesized sulfonylcarboximidamides 1–13 exhibited very low tuberculostatic activity. Minimumal inhibitory concentration (MIC) values for all the tested compounds ranged from 25 to 100 μg/cm3. No significant differences in compounds’ activity against the sensitive strain 192 and the resistant strain 210 have been observed. Isoniazid, the reference tuberculostatic, exhibited much higher activity with the MIC value 0.5–1.1 μg/cm3. These results classify the compounds tested as practically inactive against M. tuberculosis.
Antibacterial and antifungal activities
Antibacterial and antifungal activities of newly synthesized compounds were also examined. In the study of antibacterial activity three recommended reference strains S. aureus ATCC 25923, E. coli ATCC 25922, and P. aeruginosae ATCC 27853 were used [37]. Antifungal activity was determined with use of two strains: C. albicans ATCC 90028 and C. parapsilosis ATCC 22019 [38]. The susceptibility of the microorganisms to the agents was determined by the broth microdilution assay according to the procedures outlined by the National Committee for Clinical Laboratory Standards [37, 38]. The stock solutions of the agents were prepared by dissolving the chemicals in DMSO. The final concentration of the agents in 200 mm3 of Mueller-Hinton broth (for bacterial strains) or in RPMI 1,640 (for fungi) ranged over 0.125–256 μg/cm3.
In order to prepare the bacterial suspension, overnight culture of bacteria in 3 % Triptic soy broth was diluted in sterile saline to the final concentration of approximately 107 CFU/cm3. Aliquots (10 mm3) of bacterial suspension were added to each agent solution. The MIC was defined as the lowest concentration of the agent that completely inhibited growth of the bacteria after 18 h incubation at 35 °C.
Inocula of candida strains were prepared by suspension of five colonies picked from 24 h old cultures on Saburaud agar in sterile saline to the concentration of 106 cells per cm3. The final concentration of the working suspension was approximately 104 cells per cm3. Aliquots (10 mm3) of the suspension were added to each agar solution. The MIC was defined as the lowest concentration of the agent that completely inhibited growth of the fungi after 48 h incubation at 35 °C. The final results were average values from two independent experiments.
The study showed no antibacterial and antifungal activity of the tested compounds. All of the synthesized sulfonylcarboximidamides 1–13 exhibited activity with MIC > 256 μg/cm3, which meant that those values did not fit standard test concentrations.
Conclusion
In conclusion, a series of novel sulfonyl-carboximidamides with different six-membered nitrogen heterocyclic systems were synthesized successfully in a reaction of heterocyclic methyl carbimidates with benzene sulfonamide and 4-aminobenzenesulfonamide. All these new compounds were confirmed by IR and NMR spectra as well as elemental analysis. The molecular structure of the obtained compounds was discussed. Compounds possessing heterocyclic systems with a nitrogen atom in the α position to the functional group showed a single-crystal structure different from expected and described for that chemical group in the literature. Antimicrobial activity of the synthesized compounds was evaluated against M. tuberculosis, S. aureus, E. coli, P. aeruginosae, C. albicans, and C. parapsilosis. Unfortunately, all of the studied compounds were practically inactive towards microbial strains tested.
Experimental
All materials and solvents were of analytical reagent grade. Thin-layer chromatography was performed on Merck silica gel 60F254 plates and visualized with UV. The results of elemental analyses (C, H, N) for all obtained compounds were in agreement with calculated values within the range of ±0.3 %. 1H NMR spectra in CDCl3 or DMSO-d 6 were recorded on Varian Unity Plus (500 MHz) and Varian Gemini (200 MHz) instruments (Varian, Palo Alto, CA). Infrared spectra were determined as KBr pellets of the solids on a Satellite FT-IR spectrophotometer (Mattson Instruments, Madison, WI). Melting points were determined with a Boethius apparatus (Franz Küstner Nachf. KG, Dresden, Germany). Methyl pyrazine-2-carbimidate and methyl 6-methoxypyrazine-2-carbimidate required for further syntheses were obtained according to the method described earlier by Foks and co-workers [22, 23].
General method for the synthesis of sulfonyl-carboximidamides 1–8
The respective carbonitrile (1 mmol) and 0.4 cm3 (2 mmol) of DBU were refluxed in 10 cm3 of methanol for 0.5 h. Then 0.8 mmol of benzene sulfonamide or 4-aminobenzenesulfonamide was added. The mixture was refluxed for another 3 h. Then methanol was evaporated in vacuo, and 30 cm3 of water was added to the residue. The precipitate of the product was filtered off, dried, and purified by recrystallization from a suitable solvent.
N′-(Phenylsulfonyl)picolinimidamide (1, C12H11N3O2S)
Recrystallization from ethanol afforded 138 mg (66 %) 1. M.p.: 165–166 °C; IR (KBr): \( \bar{\nu } \) = 3,432, 3,320 (ν N–H), 1,613, 1,538 (ν C = C), 1,280, 1,147 (ν SO2), 757 (γ C–H), 688 (γ N–H), 589, 557 cm−1; 1H NMR (200 MHz, CDCl3): δ = 7.43–7.59 (m, 4H, 3H Ph and 1H NH + D2O exchangeable), 7.82 (m, 1H, pyridine), 8.02 (m, 2H, Ph), 8.28 (m, 2H, pyridine), 8.33 (brs, 1H, NH + D2O exchangeable), 8.58 (m, 1H, pyridine) ppm; 13C NMR (50 MHz, CDCl3): δ = 123.10 (C-3), 126.38 (C-2′, C-6′), 127.82 (C-5), 129.32 (C-3′, C-5′), 132.71 (C-4′), 138.40 (C-4), 142.39 (C-1′), 148.71 (C-6), 149.20 (C-2), 159.20 (C = N) ppm.
N′-(4-Aminophenylsulfonyl)picolinimidamide (2, C12H12N4O2S)
Recrystallization from dioxane afforded 124 mg (56 %) 2. M.p.: 202–205 °C; IR (KBr): \( \bar{\nu } \) = 3,435, 3,400, 3,323, 3,253 (ν N–H), 1,610, 1,588 (ν C = C), 1,271, 1,144 (ν SO2), 1,091 (δ C–H), 821 (γ C–H), 566 (γ N–H) cm−1; 1H NMR (200 MHz, CDCl3): δ = 5.94 (s, 2H, NH2 + D2O exchangeable), 6.52 (d, 2H, Ph, J = 8.6 Hz), 7.57 (d, 2H, Ph, J = 8.8 Hz), 7.63–7.67 (m, 1H, pyridine), 7.93–8.11 (m, 3H, 2H pyridine and 1H NH + D2O exchangeable), 8.67 (d, 1H, pyridine, J = 4.8 Hz), 8.84 (brs, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 112.79 (C-3′, C-5′), 122.83 (C-3), 127.57 (C-5), 128.38 (C-2′, C-6′), 138.26 (C-4, C-1′), 149.08 (C-6, C-4′), 152.93 (C-2), 158.04 (C = N) ppm.
N′-(Phenylsulfonyl)nicotinimidamide (3, C12H11N3O2S)
Recrystallization from dioxane–methanol mixture (1:1) afforded 98 mg (47 %) 3. M.p.: 176–178 °C; IR (KBr): \( \bar{\nu } \) = 3,439, 3,322 (ν N–H), 3,054 (ν C–H), 1,618, 1,518 (ν C = C), 1,274, 1,164, 1,149 (ν SO2), 825, 789 (γ C–H), 583 (γ N–H), 561 cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 7.47–7.66 (m, 4H, Ph), 7.94–7.98 (m, 2H, Ph), 8.16–8.20 (m, 1H, pyridine), 8.30–8.60 (brs, 1H, NH + D2O exchangeable), 8.73–8.75 (m, 1H, pyridine), 8.98 (d, 1H, pyridine, J = 1.47 Hz), 9.10–9.40 (brs, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 123.71 (C-4), 125.83 (C-2′, C-6′), 126.40 (C-3′, C-5′), 129.67 (C-3), 132.06 (C-4′), 132.56 (C-4), 135.99 (C-1′), 148.96 (C-2), 153.05 (C-6), 161.39 (C = N) ppm.
N′-(4-Aminophenylsulfonyl)nicotinimidamide (4, C12H12N4O2S)
Recrystallization from dioxane–ethanol mixture (1:1) afforded 132 mg (60 %) 4. M.p.: 215–217 °C; IR (KBr): \( \bar{\nu } \) = 3,448, 3,394, 3,337, 3,313, 3,248 (ν N–H), 2,923, 2,851 (ν C–H), 1,643, 1,612, 1,591, 1,528 (ν C = C), 1,269, 1,141 (ν SO2), 1,089 (δ C–H), 786, 698 (γ C–H), 562 (γ N–H) cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 5.92 (s, 2H, NH2 + D2O exchangeable), 6.58 (d, 2H, Ph, J = 8.7 Hz), 7.46–7.52 (m, 1H, pyridine), 7.58 (d, 2H, Ph, J = 8.7 Hz), 8.12–8.18 (m, 2H, 1H pyridine and 1H NH + D2O exchangeable), 8.70–8.73 (m, 1H, pyridine), 8.95 (d, 1H, pyridine, J = 1.9 Hz), 9.02 (s, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 112.76 (C-3′, C-5′), 123.68 (C-5), 127.81 (C-3), 128.39 (C-2′, C-6′), 129.86 (C-1′), 135.84 (C-2, C-4), 148.85 (C-6), 152.82 (C-4′), 160.13 (C = N) ppm.
N′-(Phenylsulfonyl)isonicotinimidamide (5, C12H11N3O2S)
Recrystallization from methanol–water mixture (1:1) afforded 111 mg (53%) 5. M.p.: 155–156 °C; IR (KBr): \( \bar{\nu } \) = 3,379 (ν N–H), 3,058, 2,925 (ν C–H), 1,644, 1,530 (ν C = C), 1,281, 1,142 (ν SO2), 1,086 (δ C–H), 843 (γ C–H), 589 (γ N–H), 556 cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 7.54–7.76 (m, 5H, Ph), 7.96 (d, 2H, pyridine, J = 6.6 Hz), 8.49 (s, 1H, NH + D2O exchangeable), 8.71 (d, 2H, pyridine, J = 5 Hz), 9.30 (brs, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 121.90 (C-3, C-5), 126.42 (C-2′, C-6′), 129.27 (C-3′, C-5′), 132.65 (C-4′), 141.26 (C-4), 142.41 (C-1′), 150.52 (C-2, C-6), 161.23 (C = N) ppm.
N′-(4-Aminophenylsulfonyl)isonicotinimidamide (6, C12H12N4O2S)
Recrystallization from dioxane–ethanol mixture (1:1) afforded 152 mg (69 %) 6. M.p.: 226–229 °C; IR (KBr): \( \bar{\nu } \) = 3,441, 3,357, 3,242 (ν N–H), 2,957, 2,849 (ν C–H), 1,644, 1,596, 1,527 (ν C = C), 1,276, 1,136 (ν SO2), 1,084 (δ C–H), 828 (γ C–H), 556 (γ N–H) cm−1; 1H NMR (500 MHz, DMSO-d 6): δ = 5.96 (s, 2H, NH2 + D2O exchangeable), 6.59 (d, 2H, J = 8.8 Hz), 7.74 (d, 2H, pyridine, J = 5.9 Hz), 8.25 (brs, 1H, NH + D2O exchangeable), 8.71 (d, 2H, pyridine, J = 5.9 Hz), 9.10 (brs, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 112.78 (C-3′, C-5′), 121.95 (C-3, C-5), 126.43 (C-2′, C-6′), 129.87 (C-1′), 141.36 (C-4), 150.48 (C-2, C-6), 152.85 (C-4′), 161.28 (C = N) ppm.
N′-(Phenylsulfonyl)pyrimidine-2-carboximidamide (7, C11H10N4O2S)
Recrystallization from dioxane afforded 107 mg (51 %) 7. M.p.: 206–208 °C; IR (KBr): \( \bar{\nu } \) = 3,396, 3,330 (ν N–H), 1,621,1,554 (ν C = C), 1,280, 1,151 (ν SO2), 833, 790, 689 (γ C–H), 590 (γ N–H), 501 cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 7.54–7.73 (4H, 3H Ph and 1H pyrimidine), 7.91 (d, 2H, Ph, J = 8.2 Hz), 8.21 (brs, 1H, NH + D2O exchangeable), 8.53 (brs, 1H, NH + D2O exchangeable), 8.96 (d, 2H, pyrimidine, J = 4.6 Hz) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 123.58 (C-5), 126.50 (C-2′, C-6′), 129.27 (C-3′, C-5′), 132.68 (C-4′), 142.39 (C-1′), 158.11 (C-4, C-6), 158.53 (C-2), 159.24 (C = N) ppm.
N′-(4-Aminophenylsulfonyl)pyrimidine-2-carboximidamide (8, C11H11N5O2S)
Recrystallization from ethylene glycol–methanol mixture (1:1) afforded 175 mg (79 %) 8. M.p.: 259–261 °C; IR (KBr): \( \bar{\nu } \) = 3,380, 3,330, 3,237 (ν N–H), 1,621, 1,592, 1,562, 1,503, 1,391 (ν C = C), 1,268, 1,142 (ν SO2), 830, 787 (γ C–H), 678, 578 (γ N–H), 546 cm−1; 1H NMR (500 MHz, DMSO-d 6): δ = 5.95 (s, 2H, NH2 + D2O exchangeable), 6.52 (d, 2H, Ph, J = 8.8 Hz), 7.54 (d, 2H, Ph, J = 8.8 Hz), 7.57 (t, 1H, pyrimidine, J = 4.8 Hz), 8.20 (brs, 1H, NH + D2O exchangeable), 8.87 (brs, 1H, NH + D2O exchangeable), 8.94 (d, 2H, pyrimidine, J = 4.8 Hz) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 112.76 (C-3′, C-5′), 123.50 (C-5), 127.51 (C-1′), 128.54 (C-2′, C-6′), 152.96 (C-4′), 157.71 (C-2), 158.09 (C-4, C-6), 158.50 (C = N) ppm.
N′-[4-[N-[Amino(pyrimidin-2-yl)methylene]sulfamoyl]phenyl]acetamide (9, C13H13N5O3S)
Method A: the title compound was obtained according to the method described above for compounds 1–8 from 0.11 cm3 (1 mmol) of 2-cyanopyrimidine and 0.43 g (2 mmol) of N-(4-sulfamoylphenyl)acetamide affording 112 mg (35 %) 9.
Method B: sulfonylcarboximidamide 8 (0.28 g, 1 mmol) was refluxed for 0.5 h in a solution of 0.5 cm3 (5 mmol) of acetic anhydride in 5 cm3 of pyridine. Then pyridine was evaporated in vacuo, and 20 g of ice was added to the residue. The precipitate was filtered off, dried, and recrystallized from ethylene glycol to afford 268 mg (84 %) 9.
M.p.: 253–254 °C; IR (KBr): \( \bar{\nu } \) = 3,385, 3,301 (ν N–H), 2,924, 2,854 (ν C–H), 1,684 (ν C = O), 1,624, 1,590, 1,562, 1,525, 1,401 (ν C = C), 1,280, 1,148 (ν SO2), 736 (γ C–H), 565 (γ N–H) cm−1; 1H NMR (500 MHz, DMSO-d 6): δ = 2.09 (s, 3H, CH3), 7.72 (t, 1H, pyrimidine, J = 4.8 Hz), 7.75 (d, 2H, Ph, J = 8.8 Hz), 7.86 (d, 2H, Ph, J = 8.8 Hz), 8.42 (brs, 1H, NH + D2O exchangeable), 8.96 (d, 2H, pyrimidine, J = 4.1 Hz), 9.08 (brs, 1H, NH + D2O exchangeable), 10.33 (s, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 24.39 (CH3), 118.73 (C-3′, C-5′), 123.58 (C-5), 127.73 (C-2′), 136.03 (C-1′), 143.08 (C-4′), 158.11 (C-4, C-6), 158.46 (C-2), 158.80 (C = N) ppm.
General procedure for the synthesis of sulfonylcarboximidamides 10–13
Methyl pyrazine-2-carbimidate or methyl 6-methoxypyrazine-2-carbimidate (3 mmol) and the respective sulfonamide (2.5 mmol) were refluxed in 5 cm3 of diglyme for 15 min. After cooling down 20 g of ice was added to the mixture, and the precipitate of the product was filtered off, dried, and purified by recrystallization from a suitable solvent with activated carbon.
N′-(Phenylsulfonyl)pyrazine-2-carboximidamide (10, C11H10N4O2S)
Recrystallization from dioxane afforded 249 mg (38 %) 10. M.p.: 218–219 °C; IR (KBr): \( \bar{\nu } \) = 3,434, 3,321 (ν N–H), 1,612, 1,545 (ν C = C), 1,278, 1,151 (ν SO2), 801, 686 (γ C–H), 590 (γ N–H) cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 7.54–7.69 (m, 3H, Ph), 7.98 (d, 2H, Ph, J = 7.3 Hz), 8.44 (brs, 1H, NH + D2O exchangeable), 8.77 (s, 1H, pyrazine), 8.90 (s, 1H, pyrazine), 9.15 (brs, 1H, NH + D2O exchangeable), 9.23 (s, 1H, pyrazine) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 126.49 (C-2′, C-6′), 129.35 (C-3′, C-5′), 132.84 (C-4′), 143.92 (C-2), 144.21 (C-3, C-5), 144.54 (C-1′), 148.49 (C-6), 158.28 (C = N) ppm.
N′-(4-Aminophenylsulfonyl)pyrazine-2-carboximidamide (11, C11H11N5O2S)
Recrystallization from dioxane afforded 381 mg (55 %) 11. M.p.: 247–249 °C; IR (KBr): \( \bar{\nu } \) = 3,431, 3,394, 3,320, 3,252 (ν N–H), 1,612, 1,593 (ν C = C), 1,268, 1,145 (ν SO2), 1,092 (δ C–H), 798 (γ C–H), 567 (γ N–H) cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 5.97 (s, 2H, NH2 + D2O exchangeable), 6.57 (d, 2H, Ph, J = 8.7 Hz), 7.59 (d, 2H, Ph, J = 8.7 Hz), 8.14 (brs, 1H, NH + D2O exchangeable), 8.75 (d, 1H, pyrazine, J = 2.4 Hz), 8.89 (d, 1H, pyrazine, J = 2.4 Hz), 8.92 (brs, 1H, NH + D2O exchangeable), 9.20 (s, 1H, pyrazine) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 112.82 (C-3′, C-5′), 128.53 (C-2′, C-6′), 143.83 (C-2), 144.02 (C-3, C-5), 144.78 (C-1′), 148.23 (C-6), 153.05 (C-4′), 157.05 (C = N) ppm.
N′-(Phenylsulfonyl)-6-methoxypyrazine-2-carboximidamide (12, C12H12N4O3S)
Recrystallization from ethanol afforded 584 mg (80 %) 12. M.p.: 156–157 °C; IR (KBr): \( \bar{\nu } \) = 3,395, 3,300 (ν N–H), 1,640, 1,580, 1,543 (ν C = C), 1,383 (δ C–H), 1,306, 1,144 (ν SO2), 1,008 (δ C–H), 803 (γ C–H), 591 (γ N–H) cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 4.03 (s, 3H, OCH3), 7.53–7.65 (m, 3H, Ph), 8.00 (d, 2H, Ph, J = 7.6 Hz), 8.50 (brs, 1H, NH + D2O exchangeable), 8.53 (s, 1H, pyrazine), 8.77 (s, 1H, pyrazine), 9.02 (brs, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 54.54 (OCH3), 126.44 (C-2′, C-6′), 129.33 (C-3′, C-5′), 132.77 (C-3), 135.61 (C-4′), 139.67 (C-2), 140.88 (C-1′), 142.24 (C-5), 158.27 (C-6), 159.01 (C = N) ppm.
N′-(4-Aminophenylsulfonyl)-6-methoxypyrazine-2-carboximidamide (13, C12H13N5O3S)
Recrystallization from methanol afforded 637 mg (83 %) 13. M.p.: 188–189 °C; IR (KBr): \( \bar{\nu } \) = 3,468, 3,417, 3,370, 3,309, 3,244 (ν N–H), 1,634, 1,584, 1,545 (ν C = C), 1,379 (δ C–H), 1,318, 1,261, 1,133 (ν SO2), 1,079 (δ C–H), 788 (γ C–H), 544 (γ N–H) cm−1; 1H NMR (200 MHz, DMSO-d 6): δ = 4.02 (s, 3H, OCH3), 5.95 (s, 2H, NH2 + D2O exchangeable), 6.59 (d, 2H, Ph, J = 8.8 Hz), 7.59 (d, 2H, Ph, J = 8.5 Hz), 8.21 (s, 1H, NH + D2O exchangeable), 8.51 (s, 1H, pyrazine), 8.73 (s, 1H, pyrazine), 8.77 (s, 1H, NH + D2O exchangeable) ppm; 13C NMR (50 MHz, DMSO-d 6): δ = 54.52 (OCH3), 112.76 (C-3′, C-5′), 128.56 (C-2′, C-6′), 132.74 (C-3), 139.84 (C-2), 141.76 (C-1′), 142.27 (C-5), 158.29 (C-6), 157.88 (C = N) ppm.
Crystal structure of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide
Single crystals of N′-(4-aminophenylsulfonyl)-4-chloropicolinimidamide suitable for X-ray diffraction were obtained from ethanol by slow evaporation of the solvent at room temperature. Good quality single-crystal specimens were selected for experiments at T = 295(2) K. They were mounted with epoxy glue at the tip of glass capillaries. Diffraction data were collected on an Oxford Diffraction Gemini R ULTRA Ruby CCD diffractometer with MoKα radiation (λ = 0.71073 Å). The lattice parameters were obtained by least-squares fit to the optimized setting angles of the collected reflections by means of CrysAlis CCD [27]. Data were reduced by using CrysAlis RED [27] software with applying multi-scan absorption corrections (empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm). The structural resolution procedure was made using the SHELXS-97 package solving the structures by direct methods and carrying out refinements by full-matrix least-squares on F 2 using the SHELXL-97 program [28]. All H atoms bound with aromatic C atoms were placed geometrically and refined using a riding model with C–H = 0.93 Å and U iso(H) = 1.2 U eq(C). All H atoms bound with N atoms were placed geometrically and refined using a riding model with N–H = 0.86 Å and U iso(H) = 1.5 U eq(N). The –NH2 group containing the N19 atom was assumed to be planar-trigonal and coplanar with the mean plane of the benzene ring. The –NH2 group containing the N16 atom was assumed to be planar-trigonal and coplanar with the mean plane delineated by C2, C7, and N8 atoms. All interactions demonstrated were found by the PLATON program [29]. The programs used to prepare molecular graphics were: ORTEPII [30], PLUTO-78 [31], and Mercury [32]. Full crystallographic details, excluding structural features, have been deposited (deposition no. 849210) with the Cambridge Crystallographic Data Center. These data may be obtained, on request, from the Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Tel.: +44-1223-336408; Fax: +44-1223-336033; e-mail:deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
References
Gomez-Lus R, Clavel A, Castillo J, Seral C, Rubio C (2000) Int J Antimicrob Agents 16:335
Appelbaum PC (2007) Clin Infect Dis 45:S165
Prasad R, Kapoor K (2004) Int Rev Cytol 242:215
Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, Jensen P, Bayona J (2010) Lancet 375:1830
Wells CD, Cegielski JP, Nelson LJ, Laserson KF, Holtz TH, Finlay A, Castro KG, Weyer K (2007) J Infect Dis 196:S68
Norrby SR, Nord CE, Finch R (2005) Lancet Infect Dis 5:115
Supuran CT (2008) Nat Rev Drug Disc 7:168
Isik S, Kockar F, Aydin M, Arslan O, Guler OO, Innocenti A, Scozzafava A, Supuran CT (2009) Bioorg Med Chem 17:1158
Gawin R, De Clercq E, Naesens L, Koszytkowska-Stawińska M (2008) Bioorg Med Chem 16:8379
Bouissane L, Kazzouli SE, Léonce S, Pfeiffer B, Rakib EM, Khouili M, Guillaument G (2006) Bioorg Med Chem 14:1078
Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, Kiebe G (2004) J Med Chem 47:550
Panico A, Vicini P, Incert M, Cardile V, Gentile B, Ronsisvalle G (2002) Il Farmaco 57:671
Sienkiewicz P, Bielawski K, Bielawska A, Palka J (2005) Environ Toxicol Pharmacol 20:118
Sielecki TM, Liu J, Mousa SA, Racanelli AL, Hausner EA, Waxler RR, Olson RE (2001) Bioorg Med Chem Lett 11:2201
Echevarria A, Santos LH, Miller J, Mahmood N (1996) Bioorg Med Chem Lett 6:1901
Bedi PMS, Mahajan MP, Kapoor VK (2004) Bioorg Med Chem Lett 14:3821
Greenidge PA, Carlsson B, Bladh L-G, Gillner M (1998) J Med Chem 41:2503
Dubina VL, Shebitchenko LN, Pedan VP, Yukhno AG, Skripets VI (1982) Russ J Org Chem 18:691
Rossi E, Stradi R, Benedusi A (1987) Tetrahedron 43:4785
Fujisawa T, Mizuno C (1952) Yakugaku Zasshi 72:694
Northey EH, Pierce AE, Kartesz DJ (1942) J Am Chem Soc 64:2763
Foks H, Janowiec M (1979) Acta Polon Pharm 36:155
Foks H, Manowska W (1976) Pol J Pharmacol Parm 28:49
Bogdanowicz A, Foks H, Kędzia A, Kwapisz E, Zwolska Z, Augustynowicz-Kopeć E (2009) Heterocycles 78:2217
Shilcrat SC, Mokhallati MK, Fortunak JMD, Pridgen LN (1997) J Org Chem 62:8449
Hopkins KT, Wilson WD, Bender BC, McCurdy DR, Hall JE (1998) J Med Chem 41:3872
Oxford Diffraction (2008) CrysAlis CCD and CrysAlis RED. Yarnton, England
Sheldrick GM (2008) Acta Cryst A64:112
Spek AL (2009) Acta Cryst D65:148
Johnson CK (1976) ORTEP II. Report ORNL-5138. Oak Ridge National Laboratory, Oak Ridge
Mortherwell S (1978) Clegg S PLUTO-78 program for drawing and molecular structure. University of Cambridge, UK
Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, van de Streek J, Wood PAJ (2008) Appl Cryst 41:466
Youmans GP (1947) Am Rev Tuberc 56:376
Youmans GP, Youmans AS (1949) J Bacteriol 58:247
Atlas RM, Singler JW (1995) Media for clinical microbiology. CRC Press, Boca Raton, p 313
Foks H, Buraczewska M, Manowska W, Sawlewicz J (1971) Dissert Pharm Pharmacol 23:49
National Committee for Clinical Laboratory Standards (1993) Methods for dilution antimicrobial susceptibility test for bacteria that grow aerobically. 3rd edn. Approved standard NCCLS document M7-A3, No 24, vol 13, NCCLS, Villanova
National Committee for Clinical Laboratory Standards (1993) Reference method for broth dilution antifungal susceptibility testing of yeast. Proposed Standard Document M27-P, Vol 13, No 24, NCCLS, Villanova
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gobis, K., Foks, H., Wiśniewska, K. et al. Synthesis, structure, and antimicrobial activity of heterocyclic phenylsulfonyl- and 4-aminophenylsulfonyl-carboximidamides. Monatsh Chem 143, 1161–1169 (2012). https://doi.org/10.1007/s00706-012-0769-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-012-0769-6