
Abstract
Rotaviruses are major causative agents of acute diarrhea in children under 5 years of age in Malaysia. However, a rotavirus vaccine has not been included in the national vaccination program. To date, only two studies have been carried out in the state of Sabah, Malaysia, although children in this state are at risk of diarrheal diseases. Previous studies showed that 16%–17% of cases of diarrhea were caused by rotaviruses and that equine-like G3 rotavirus strains are predominant. Because the prevalence of rotaviruses and their genotype distribution vary over time, this study was conducted at four government healthcare facilities from September 2019 through February 2020. Our study revealed that the proportion of rotavirus diarrhea increased significantly to 37.2% (51/137) after the emergence of the G9P[8] genotype in replacement of the G12P[8] genotype. Although equine-like G3P[8] strains remain the predominant rotaviruses circulating among children, the Sabahan G9P[8] strain belonged to lineage VI and was phylogenetically related to strains from other countries. A comparison of the Sabahan G9 strains with the G9 vaccine strains used in the RotaSiil and Rotavac vaccines revealed several mismatches in neutralizing epitopes, indicating that these vaccines might not be effective in Sabahan children. However, a vaccine trial may be necessary to understand the precise effects of vaccination.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Globally, rotaviruses continue to be the major causative agents of diarrhea in children under 5 years of age [1], with > 95% of the mortality associated with this condition occurring in sub-Saharan Africa, South Asia, and Southeast Asia [2,3,4,5]. Rotaviruses accounted for 40% of all diarrhoea cases in Southeast Asia from 1990 to 2017 [1]. In Malaysia, the burden of rotaviruses remains unclear, as there is a considerable gap in surveillance studies, with the exception of the 2002–2010 period [6]. This is notable for Sarawak and Sabah, which are two states of Malaysian Borneo where indigenous people of different ethnicities reside. Although, indigenous children in Malaysian Borneo are more vulnerable to acute gastroenteritis than those in other localities [7] in Sabah, only two studies have been carried out, during 2005–2006 and 2018–2019, which showed that rotaviruses are responsible for 16%–17% of all diarrhea cases [8, 9].
Rotaviruses are double-stranded RNA viruses whose genome is divided into 11 segments. Gene segments 1, 2, 3, 4, 6, and 9 encode the structural proteins VP1, VP2, VP3, VP4, VP6, and VP7, respectively, whereas gene segments 5, 8, 7, 10, and 11 encode the non-structural proteins NSP1, NSP2, NSP3, NSP4, and NSP5/6, respectively [10]. Trypsin treatment results in the specific cleavage of VP4 to form VP8* and VP5*, representing the amino- and carboxyl-terminal regions of the protein, respectively. The viral genome is enclosed by an icosahedral capsid consisting of an inner core, an intermediate capsid, and an outer capsid. The VP4 and VP7 proteins are the main components of the outer capsid and contain epitopes that induce neutralizing antibodies and define the P and G genotypes, respectively, of the dual nomenclature. To date, 42 G and 58 P genotypes have been discovered in humans and animals [https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/newgenotypes (accessed October 12, 2022)]. Due to the segmented nature of their genome, rotaviruses can be characterized according to electropherotype patterns based on the differences in the relative migration rates of genomic segments in polyacrylamide gel electrophoresis (PAGE), and this is useful for detection of strain diversification. Moreover, also because of the segmented nature of the rotavirus genome, gene reassortment occurs between human and animal rotaviruses, resulting in rotavirus strain diversity [11]. Point mutations in genes encoding outer capsid proteins are another evolutionary mechanism that can be used to classify rotaviruses into lineages, and some of these result in the emergence of antibody-escape mutants [12].
Four oral live-attenuated commercial vaccines, i.e., Rotarix, RotaTeq, Rotavac, and RotaSiil, have been prequalified by the World Health Organization. Rotarix (GlaxoSmithKline Biologicals SA, Rixensart, Belgium) is a monovalent vaccine consisting of the G1P[8] genotype, whereas RotaTeq (Merck & Co., Inc., West Point, PA, USA) is a pentavalent vaccine that includes the G1–G4 and P[8] genotypes. India has introduced its own vaccines, i.e., Rotavac (Bharat Biotech Telangana, India), a monovalent vaccine carrying the G9P[11] genotype, and RotaSiil (Serum Institute of India, Pune, India), which is a pentavalent vaccine containing the G1, G2, G3, G4, and G9 genotypes [13]. Despite a decrease in the rotavirus burden since the implementation of vaccination, most of the developing countries in Asia do not include rotavirus vaccination in their national vaccination program [1]. Malaysia is not an exception compared with other Asian countries, but Rotarix and RotaTeq are available in the private sector [9].
The predominant rotavirus types worldwide affecting human populations are G1P[8], G3P[8], G4P[8], G9P[8], and G2P[4], which have been identified in Malaysia, albeit with varying prevalence [6]. The predominant circulating genotypes change and vary according to location, which is very important, as vaccine development is based on the predominant circulating strains. Because the genotypes change, continued surveillance is important to evaluate the effectiveness of the rotavirus vaccines used in the national vaccination program. The genotype distribution of rotavirus in Sabah was first assessed in 2018–2019 [9]. In the present follow-up study (2019–2020), we detected an increase of rotavirus infection, which was associated with the emergence of G9 strains over a short time span.
Materials and methods
Ethical approval
Ethical approval was obtained from the Medical Research Ethics Committee, Ministry of Health, Malaysia, for Menggatal Health Clinic and Telipok Health Clinic (NMRR-16-2245-32787), Sabah Women and Children Hospital (NMRR-19-3925-52370), and Kunak District Hospital (NMRR-20-1324-55178). All procedures were performed in accordance with relevant guidelines and regulations. Informed consent was obtained from the legal guardians of the children participating in this study.
Collection of stool samples and patients’ information
The study period was from September 2019 through February 2020. Stool samples were collected from children aged ≤5 years who attended the above-mentioned healthcare facilities with watery diarrhea with or without vomiting and fever. Watery diarrhea was defined as passing watery stools at least three times during a 24-h period. Patient information such as gender, age in months, and ethnicity was recorded. Stool samples were collected at healthcare facilities in stool-collection bottles, diluted in phosphate-buffered saline (PBS) to make 10% suspensions, transported under cold-chain conditions to Universiti Malaysia Sabah, and stored at -80°C until use.
Rotavirus detection and genomic extraction of RNA
Rotavirus was detected using a commercial enzyme immunoassay kit according to the manufacturer’s instructions (Rotaclone, Meridien Diagnostics Inc., Cincinnati, USA). Rotavirus genomic RNA was extracted from rotavirus-positive samples using a QIAamp Viral RNA Mini Kit according to the manufacturer’s instructions (QIAGEN GmbH, Hilden, Germany). For electropherotyping, genomic RNA was extracted using the phenol:chloroform:isoamyl alcohol method [9].
Determination of G and P genotypes by RT-PCR
For rotavirus VP7 and VP4 gene amplification, reverse transcription (RT-PCR) was performed using an AccessQuick RT-PCR (Promega Corporation, Madison, WI, USA) according to the instructions [14]. The VP7 gene was amplified using the primers Beg9 and End9 [15], and the VP4 gene was amplified using the primers Con2 and Con3 [16]. For G and P genotyping, multiplex PCR was performed using a master mix (Promega Corporation, Madison, WI, USA). Primers for the detection of the G1, G2, G3, G4, G8, G9, G10, and G12 genotypes were used as described previously [15, 18]. For P genotyping, primers for the detection of P[4], P[6], P[8], P[9], P[10], and P[11] were used [17, 18]. The nucleotide sequences of the VP7 (1,062 bp) and VP8* (876 bp) regions of the VP4 gene were determined using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). The sequencing products were analyzed using an ABI Prism 3100 Genetic Analyzer (Applied Biosystems).
Phylogenetic analysis
The VP7 and VP8* regions of the VP4 gene were sequenced as described previously [14]. Similar sequences were identified using BLAST (www.ncbi.nlm.nih.gov/blast), multiple sequence alignment was performed using ClustalW, and phylogenetic trees were constructed by the maximum-likelihood method using MEGA X software [19]. Bootstrap analysis of 1,000 replicates was used to investigate the significance of the branching of the constructed trees. The nucleotide sequence identity values for individual genes were calculated using online software (www.bioinformatics.org).
Determination of electropherotypes
Electropherotypes were determined by polyacrylamide gel electrophoresis (PAGE) of the extracted dsRNA of rotavirus-positive samples [9]. In brief, 5 µl of extracted dsRNA was mixed with 5 µl of loading buffer, loaded onto a 10% polyacrylamide gel, and subjected to electrophoresis for 16 h at a constant current of 8 mA. The gel was then stained with ethidium bromide to visualize the migration pattern of the genomic RNA. Electropherotype numbers were assigned arbitrarily based on differences in the migration patterns within at least one of the four groups of segments, i.e., segments 1 to 4, 5 and 6, 7 to 9, or 10 and 11.
Comparison of the VP7 antigenic epitopes of circulating rotaviruses with those of vaccine strains
To compare the antigenic epitopes on the VP7 protein of the G9 rotaviruses circulating in Sabah with those of the Rotavac (G9P[11]) and RotaSiil (G9P[5]) vaccine strains, multiple sequence alignments of the amino acid sequences of VP7 were made using the ClustalW plug-in in MEGA X software [19].
Statistical analysis
To assess whether children with G9 vs. those with non-G9 genotypes differed regarding age at presentation, an independent two-tailed t-test was performed, using Social Science Statistics software (https://www.socscistatistics.com/tests/studentttest/default.aspx). A P-value less than 0.05 was considered statistically significant.
Results
Rotavirus prevalence and age distribution
A total of 137 watery stool samples were collected from Sabah Women and Children Hospital (78.1%; 107/137), Kunak District Hospital (13.9%; 19/137), Menggatal Health Clinic (5.8%; 8/137), and Telipok Health Clinic (2.2%; 3/137). The male-to-female ratio was 1.4:1 (80 males, 57 females). Children aged 12–23 months had the highest incidence of acute diarrhea (29.9%; 41/137), followed by children aged 6–11 months (28.5%; 39/137). The median age of patients with acute diarrhea was 15 months (range, 1–64 months).
Fifty-one (37.2%) of the 137 samples were positive for rotavirus. Most infections were reported at Sabah Women and Children Hospital (84.3%; 43/51), followed by Kunak District Hospital (9.8%; 5/51) and Menggatal Health Clinic (5.9%; 3/51). The male-to-female ratio was 1.4:1 (30 males, 21 females). Children aged 12–23 months had the highest incidence of rotavirus infection (33.3%; 17/51).
Genotype distribution
Of the 51 rotavirus-positive samples, 42 (82.3%) underwent successful G and P genotyping. The predominant G genotype was G3 (n = 21), followed by G9 (n = 14), G1 (n = 3), G2 (n = 1), and an undetermined Gx genotype (n = 3). “Undetermined” indicates that no primary PCR product was available. The P[8] genotype was detected in 38 rotavirus-positive samples. In four samples, the P genotype could not be determined, and these were designated P[x]. Based on the combination of the G and P genotypes, the predominant combined genotype was G3P[8] (n = 19; 45.2%), followed by G9P[8] (n = 14; 33.3%), G1P[8] (n = 2; 4.8%), GxP[8] (n = 3; 7.1%), G3P[x] (n = 2; 4.8%), G1P[x] (n = 1; 2.4%), and G2P[x] (n = 1; 2.4%) (Table 1).
The age difference between the children who were found to have the G9 genotype (14 children; mean age = 22.4 months) and those with a non-G9 genotype (26 children; mean age = 16.9 months) was not statistically significant (P = 0.111981).
Electropherotypes
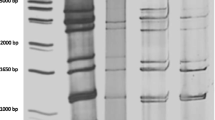
In electropherotype analysis, all 11 segments were visible in 30 out of 51 (58.8%) samples tested. One short and four long electropherotype patterns were identified (Fig. 1). The short electropherotype S1 was found in 13 isolates. Among the long electropherotypes, the L1 group included 13 isolates, the L2 and L4 groups each included one isolate, and the L3 group included two isolates. The G3P[8] genotype isolates included both short (S1) and long (L2) electropherotypes. All G9P[8]-positive samples exhibited long electropherotype patterns (L1). The G genotype could not be determined for the L3 and L4 electropherotypes.

Electropherotypes of rotaviruses identified in Sabah. In total, four electropherotypes were identified, four long (L1–L4) and one short (S1) electropherotype patterns were identified. L1 and L2 were detected in genotype G9 and G3 strains, respectively. L3 and L4 were detected in untypable strains. S1 was identified in G3 strains. Images of the original gels are presented in Supplementary Figure S1
Phylogenetic analysis
The VP7 and the VP8* portion of the VP4 gene could be sequenced in 36 and 34 isolates, respectively. Phylogenetic analysis based VP7 genes of the G1 genotype, Sabahan G1P[8] belonged to lineage Ia [20] and clustered with a significant bootstrap value with strains from Sabah detected in a previous study [6] and with strains from Indonesia detected in 2017 and 2018 (Fig. 2). It is worth mentioning that most of the G1 strains detected in the previous study from Sabah belonged to lineage II. The G2 isolate belonged to lineage VI and was related to strains from Italy, Russia, Bangladesh, Australia, and Vietnam from 2005 to 2011 (Fig. 3).

Phylogenetic tree based on nucleotide sequences of the VP7 gene of G1 strains. The human rotavirus strain L116 (genotype G3) was used as an outgroup. Numbers at nodes represent the bootstrap value, and values higher than 70% are shown. The scale bar shows the genetic distance, expressed as nucleotide substitutions per site. Strains from the present study (indicated by a filled circle) belong to lineage Ic and are clustered with strains detected in Sabah in a previous study. The GenBank accession number is shown before each strain name

Phylogenetic tree based on the VP7 gene of G2 strains. The human rotavirus strain L116 (genotype G3) was used as an outgroup. Numbers at nodes represent the bootstrap value, and values higher than 70% are shown. The scale bar shows the genetic distance, expressed as nucleotide substitutions per site. The strain identified in this study (indicated by a filled circle) belongs to lineage IV. The GenBank accession number is shown before each strain name
Four G3P[8] strains belonged to lineage III and formed an independent cluster with significant bootstrap values (Fig. 4). These strains were related to strains from China and those detected previously in Sabah. The current Sabahan strains shared 99.1–99.5% nucleotide sequence identity with each other and shared 96.7–98.3% nucleotide sequence identity with previously detected Sabahan G3P[8] strains. An additional 15 G3P[8] strains belonged to a cluster formed by the previously detected equine-like G3P[8] from Sabah and belonged to lineage I. The current Sabahan strains shared 97.5–100% nucleotide sequence identity with each other and shared 96.6–99.8% nucleotide sequence identity with previously detected Sabahan equine-like G3P[8] strains.

Phylogenetic tree based on the VP7 gene of G3 strains. The human rotavirus strain Wa (genotype G1) was used as an outgroup. Numbers at nodes represent the bootstrap value, and values higher than 70% are shown. The scale bar shows the genetic distance, expressed as nucleotide substitutions per site. The strains identified in this study belong to lineages I and III (indicated by a filled circle). The GenBank accession number is shown before each strain name
All G9P[8] strains identified in this study belonged to lineage VI and clustered together with strains from China (Hubei, Liaoning, and Shandong), Japan, Russia, Vietnam, Thailand, South Africa, and the USA (2013–2019) (Fig. 5).

Phylogenetic tree based on the VP7 gene of G9 strains. The human rotavirus strain DS-1 (genotype G2) was used as an outgroup. Numbers at nodes represent the bootstrap value, and values higher than 70% are shown. The scale bar shows the genetic distance, expressed as nucleotide substitutions per site. Strains from the present study (indicated by a filled circle) belong to lineage VI. The GenBank accession number is shown before each strain name
The VP4 genes of all Sabahan G3P[8], G1P[8], GxP[8], and G9P[8], strains belonged to lineage III and formed three clusters in the phylogenetic tree (Fig. 6). The VP4 gene of seven equine-like G3P[8], two GxP[8], one G1P[8], and all G9P[8] strains clustered with equine-like G3 strains from our previous study, with a significant bootstrap value [9]. They belonged to sublineage III.1. Another cluster contained the VP4 genes of two equine-like G3 strains and one G1[P8] strain from the present study and two G1P[8] strains from our previous study [9]. In the third cluster, the VP4 genes of three G3 and one equine-like G3 strains grouped together. The last two clusters belonged to sublineage III.4.

Phylogenetic tree based on the VP4 gene of P[8] strains. The human rotavirus strain 366 (genotype P[4]) was used as an outgroup. Numbers at nodes represent the bootstrap value, and values higher than 70% are shown. The scale bar shows the genetic distance expressed as nucleotide substitutions per site. All strains from the present study (indicated by a filled circle) belong to lineage III (sublineages III.1 and III.4). The GenBank accession number is shown before each strain name
Comparison of VP7 antigenic epitopes of Sabahan G9 strains with Rotavac and RotaSiil vaccine strains
We compared the amino acid sequences of the VP7 proteins of Sabahan G9 strains with those of the Rotavac and RotaSiil vaccine strains to identify possible epitopes of vaccine-escape strains (Fig. 7). Among the 29 amino acid residues comprising the VP7 antigenic epitopes, four residues differed from Rotavac, and three differed from RotaSiil [21]. In comparison to Rotavac, two substitutions (I87T and G100N) occurred in the 7-1a epitope, whereas no substitutions were detected in the 7-1b epitope. Moreover, two substitutions (N145D and N221S) were detected in the 7-2 epitope. In comparison to RotaSiil, two substitutions (A87T and D100N) occurred in the 7-1a epitope, one substitution (T242N) occurred in the 7-1b epitope, and no substitutions were detected in the 7-2 epitope.

Amino acid sequence alignment of the VP7 proteins of the vaccine strains Rotavac and RotaSiil with Sabahan G9 strains reported in this study. Antigenic epitopes are indicated above the residue numbers. Residues that differed from Rotavac are highlighted in green. Residues that differed from RotaSiil are highlighted in blue. Residues that differed from both Rotovac and RotaSiil are highlighted in yellow. Amino acid substitutions that have been shown to be associated with escape from neutralization by monoclonal antibodies are indicated by an asterisk (*)
Discussion
Fluctuations in the distribution of the predominant rotavirus genotypes have been reported worldwide [18, 22,23,24,25,26]. These fluctuations may be driven by herd immunity, and the emergence of a particular genotype may reflect the presence of a sufficiently large population of susceptible children [22]. One of the significant findings of this study was that, within 5 months of the end of the previous study [9], the prevalence of rotavirus infection had increased by about 256%. In this study, we found that the incidence of rotavirus infection was highest in children 12–23 months of age, as has been reported previously [27,28,29]. However, in contrast to other studies [30, 31] in which older children were more likely to be infected with G9 strains, in our study, the average age of Sabahan children infected with G9 strains was not significantly different from that of children infected with more-common strains. It is therefore likely that these children had no immunity against the newly circulating G9 strains and, when exposed, were infected.
In the present study, G3P[8] was the predominant circulating genotype among Sabahan children, followed by G9, G1, and G2. Equine-like G3P[8] possibly adapted well to the child population of Sabah, and this is supported by phylogenetic analysis showing clustering of previous and current strains. A similar observation was made for G1P[8], although this is not a predominant strain in Sabah; in contrast, previous strains adapted well and continued to infect children, as found in the present study. The most noteworthy finding was the emergence of G9 strains replacing the G12 strain that was in circulation previously (Fig. 8); moreover, G9 strains might be responsible for the abrupt rise in rotavirus infections in children compared with the previous study [9].

Rotavirus G and P genotypes found in a previous study by Amit et al., 2021 [9] (a) and in the current study (b). In both studies, G3P[8] was the predominant genotype. The present study shows the emergence of G9 strains replacing the G12 strain that was in circulation previously
Several publications have reported the emergence of the G9 genotype in other countries during approximately the same period. A study performed in 2018 in the West Nusa Tenggara region of neighboring Indonesia detected the presence of G9P[8] strains; however, these strains were absent in South Sumatra and West Papua [32], indicating a localized emergence of this genotype. Furthermore, a study carried out in 2017–2018 in China showed G9P[8] to be the predominant strain [33]. In Argentina, G9P[4] was in circulation in 2017, but not in 2018 [34]. In Thailand, G9P[8] was predominant during 2018 but decreased significantly in 2019, when G3 became predominant. Phylogenetically, Sabahan G9 was more closely related to strains circulating in three provinces of China than to those in neighboring Indonesia or Thailand, suggesting that transmission is occurring between China and Sabah state, perhaps because this state is a popular destination for Chinese tourists.
G9 was first identified in 1987 in the USA [35], and it continued to compete with other genotypes until it become the fifth globally predominant rotavirus [36]. Several studies performed in other countries have shown that G9 initially had a low prevalence but then rapidly increased to become the predominant genotype [37,38,39]. In Malaysia, G9P[8] rotavirus was detected in the early 2000s in Johor, Kuala Lumpur [40], and Sarawak [41, 42]. In Sarawak, rotavirus surveillance studies were conducted from 2001 to 2007, with G9[P8] being the predominant genotype from 2001 to 2003 and G1P[8] being predominant in 2001 and 2007 [41, 42]. The nucleotide sequences of these G9 strains from Malaysia are not available in public databases; therefore, they could not be compared with the G9 strains detected in the present study. Furthermore, for the last 10 years, except in one study [9], no genotyping of rotavirus strains has been performed in Malaysia; therefore, the dynamics of G9 distribution remain unknown. Thus, a regular surveillance system should be established to track the genotype distribution, which will be helpful for planning vaccination policy. Although the VP7 and VP4 genes of Sabahan G9 strains clustered together phylogenetically, these strains harbored four different long electropherotype patterns, indicating diversity within the G9 genotype.
The most prevalent G9 lineages worldwide are lineages III and VI [43]. Geographically, lineage VI is widespread in Asia and was identified in 2011–2019 in Tokyo (2017–2018) [44], Vietnam (2016–2018) [45], Wuhan (2019) [46], and Beijing (2011–2013). Lineages I, II, IV, and V were detected only in samples from humans, whereas lineages III and VI are epidemiologically linked to porcine and human samples [47]. Because all Sabahan G9P[8] strains belonged to lineage VI, additional studies using whole-genome sequence analysis are required to obtain a full picture of these strains regarding their origin, evolution, and reassortment. The only G2 strain detected was phylogenetically related to strains circulating in 2005–2011 in Italy, Russia, Bangladesh, Australia, and Vietnam. Why such an old strain was circulating in Sabah is puzzling and warrants further investigation. Our results indicate that the dynamics of rotavirus genotype changes in Sabah were very rapid. Other studies have shown that fluctuations in the rotavirus genotype distribution occurred continuously over time and according to location.
To shed light on vaccine effectiveness, we compared the antigenic epitopes of our G9 strains with those of the Rotavac and RotaSiil vaccine strains and identified several substitutions. There were two defined antigenic epitopes, 7-1 (7-1a and 7-1b) and 7-2 in VP7, where amino acid substitution could reduce the effectiveness of rotavirus vaccines. Epitopes 7-1a, 7-1b, and 7-2 include the earlier-described antigenic regions A (87–101) and D (291); C (208–221), E (189–190), and F (233–242); and B (142–152), respectively [48]. In this study, we found that the Rotavac and RotaSiil vaccines might have reduced effectiveness against Sabahan G9 rotaviruses, because all of them exhibited substitutions at residues that are associated with neutralization antibody escape (positions 87, 100, 145, 221 and 242) [48]. The substitution N145D results in the loss of a glycosylation site at amino acid residues 145–147 within the 7-2 epitope, making the virus resistant to neutralization [48]. The substitution N221S in this epitope can increase resistance to neutralization by tenfold [49]. The Rotavac and RotaSiil vaccines are currently in use only in India and Palestine (Rotavac only) [50].
The impact of amino acid substitutions within antigenic epitopes on vaccine effectiveness is difficult to predict, as the effectiveness of vaccines in a region can be influenced by multiple factors, ranging from concurrent enteric infections, malnutrition, immune status, healthcare access, and the vaccine coverage rate within the population [51]. Therefore, a vaccine trial may be necessary to evaluate the effectiveness of vaccination. A limitation of this study was that we had to discontinue the study earlier than scheduled because of the introduction of a movement control order (MCO) by Malaysian authorities as of March 2020 to control the spread of COVID-19. Moreover, the hospitals that participated in our study were then designated as COVID-19 hospitals, and sample collection was no longer possible. Nevertheless, the results of this study can be used to evaluate the after-effects of the MCO on rotavirus infections, considering that significant decreases in rotavirus infection were reported in Japan [52], Germany [53], and Korea [54] during the COVID-19 pandemic.
Conclusion
In this study, we found that the emergence of four different electropherotype patterns of genotype G9 in replacement of G12 was associated with a higher prevalence of rotavirus diarrhoea in children in Sabah, Malaysian Borneo; however, equine-like G3P[8] rotavirus continued to be the predominant strain in circulation.
Data availability
The datasets generated and/or analysed during the current study are available in the DNA Data Bank of Japan/European Molecular Biology Laboratory/GenBank repository: OM928419, OM928420, OM928421, OM928422, OM928423, OM928424, OM928425, OM928426, OM928427, OM928428, OM928429, OM928430, OM928431, OM928432, OM928433, OM928434, OM928435, OM928436, OM928437, OM928438, OM928439, OM928440, OM928441, OM928442, OM928443, OM928444, OM928445, OM928446, OM928447, OM928448, OM928449, OM928450, OM928451, OM928452, OM928453, OM928454, OM928455, OM928456, OM928457, OM928458, OM928459, OM928460, OM928461, OM928462, OM928463, OM928464, OM928465, OM928466,OM928467, OM928468, OM928469, OM928470, OM928471, OM928472, ON922858, ON922858, ON922859, ON922860, ON922861, ON922862, ON922863, ON922864, ON922865, ON922866, ON922867, OP004804, OP004805, OP004806, OP004807, OP004808, OP004809.
References
Lestari FB, Vongpunsawad S, Wanlapakorn N, Poovorawan Y (2020) Rotavirus infection in children in Southeast Asia 2008–2018: disease burden, genotype distribution, seasonality, and vaccination. J Biomed Sci 27(1):66
Troeger C, Khalil IA, Rao PC, Cao S, Blacker BF, Ahmed T et al (2018) Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr 172(10):958–965
Omatola CA, Olaniran AO (2022) Rotaviruses: from pathogenesis to disease control-a critical review. Viruses 14(5):875
Burnett E, Parashar UD, Tate JE (2021) Rotavirus infection, illness, and vaccine performance in malnourished children: a review of the literature. Pediatr Infect Dis J 40(10):930–936
Varghese T, Kang G, Steele AD (2022) Understanding rotavirus vaccine efficacy and effectiveness in countries with high child mortality. Vaccines (Basel) 10(3):346
Lee WS, Lim BT, Chai PF, Kirkwood CD, Lee JK (2012) Rotavirus genotypes in Malaysia and universal rotavirus vaccination. Hum Vaccin Immunother 8(10):1401–1406
Hsu VP, Abdul Rahman HB, Wong SL, Ibrahim LH, Yusoff AF, Chan LG et al (2005) Estimates of the burden of rotavirus disease in Malaysia. J Infect Dis 192(Suppl 1):S80–S86
Goh CT, Cheah PK, Soo TL, Lee WS (2009) The epidemiology and burden of childhood rotavirus infection in a tertiary hospital in Sabah, Malaysia. Med J Malaysia 64(2):146–149
Amit LN, Mori D, John JL, Chin AZ, Mosiun AK, Jeffree MS et al (2021) Emergence of equine-like G3 strains as the dominant rotavirus among children under five with diarrhea in Sabah, Malaysia during 2018–2019. PLoS ONE 16(7):e0254784
Desselberger U (2014) Rotaviruses. Virus Res 190:75–96
Yahiro T, Takaki M, Chandrasena TGAN, Rajindrajith S, Iha H, Ahmed K (2018) Human-porcine reassortant rotavirus generated by multiple reassortment events in a Sri Lankan child with diarrhea. Infect Genet Evol 65:170–186
Luchs A, TimenetskyMdo C (2016) Group A rotavirus gastroenteritis: post-vaccine era, genotypes and zoonotic transmission. Einstein (Sao Paulo) 14(2):278–287
Skansberg A, Sauer M, Tan M, Santosham M, Jennings MC (2021) Product review of the rotavirus vaccines ROTASIIL, ROTAVAC, and Rotavin-M1. Hum Vaccin Immunother 17(4):1223–1234
Mitui MT, Chandrasena TN, Chan PK, Rajindrajith S, Nelson EA, Leung TF et al (2012) Inaccurate identification of rotavirus genotype G9 as genotype G3 strains due to primer mismatch. Virol J 9:144
Gouvea V, Glass RI, Woods P, Taniguchi K, Clark HF, Forrester B et al (1990) Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J Clin Microbiol 28(2):276–282
Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, Flores J et al (1992) Identification of group A rotavirus gene 4 types by polymerase chain reaction. J Clin Microbiol 30(6):1365–1373
Gunasena S, Nakagomi O, Isegawa Y, Kaga E, Nakagomi T, Steele AD et al (1993) Relative frequency of VP4 gene alleles among human rotaviruses recovered over a 10-year period (1982–1991) from Japanese children with diarrhea. J Clin Microbiol 31(8):2195–2197
Reju S, Srikanth P, Selvarajan S, Thomas RK, Barani R, Amboiram P et al (2022) A shift in circulating rotaviral genotypes among hospitalized neonates. Sci Rep 12(1):2842
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549
Pradhan GN, Chitambar SD (2018) Full genomic analysis of G1p[8] rotavirus strains recovered from rotavirus vaccinated and non-vaccinated children hospitalized for acute gastroenteritis in Pune, Western India. J Med Virol 90(4):772–778. https://doi.org/10.1002/jmv.25007
Raju B, Parikh RP, Vetter VV, Kolhapure S (2019) Epidemiology of rotavirus gastroenteritis and need of high rotavirus vaccine coverage with early completion of vaccination schedule for protection against rotavirus diarrhea in India: a narrative review. Indian J Public Health 63(3):243. https://doi.org/10.4103/ijph.ijph_307_18
Mladenova Z, Korsun N, Geonova T, Iturriza-Gómara M, Rotavirus Study Group (2010) Molecular epidemiology of rotaviruses in Bulgaria: annual shift of the predominant genotype. Eur J Clin Microbiol Infect Dis 29(5):555–562
Manjate F, João ED, Chirinda P, Garrine M, Vubil D, Nobela N et al (2022) Molecular epidemiology of rotavirus strains in symptomatic and asymptomatic children in Manhiça district, Southern Mozambique 2008–2019. Viruses 14(1):134
Okitsu S, Khamrin P, Hikita T, Thongprachum A, Pham NTK, Hoque SA et al (2022) Changing distribution of rotavirus A genotypes circulating in Japanese children with acute gastroenteritis in outpatient clinic, 2014–2020. J Infect Public Health 15(7):816–825
Lu L, Zhong H, Jia R, Su L, Xu M, Cao L et al (2022) Prevalence and genotypes distribution of group A rotavirus among outpatient children under 5 years with acute diarrhea in Shanghai, China, 2012–2018. BMC Gastroenterol 22(1):217
Esona MD, Gautam R, Katz E, Jaime J, Ward ML, Wikswo ME et al (2021) Comparative genomic analysis of genogroup 1 and genogroup 2 rotaviruses circulating in seven US cities, 2014–2016. Virus Evol 7(1):veab023
Nirwati H, Hakim MS, Aminah S, Dwija IBNP, Pan Q, Aman AT (2017) Identification of rotavirus strains causing diarrhoea in children under five years of age in Yogyakarta, Indonesia. Malays J Med Sci 24(2):68–77
Zaraket R, Salami A, Bahmad M, El Roz A, Khalaf B, Ghssein G et al (2020) Prevalence, risk factors, and clinical characteristics of rotavirus and adenovirus among Lebanese hospitalized children with acute gastroenteritis. Heliyon 6(6):e04248
Tian Y, Chughtai AA, Gao Z, Yan H, Chen Y, Liu B et al (2018) Prevalence and genotypes of group A rotavirus among outpatient children under five years old with diarrhea in Beijing, China, 2011–2016. BMC Infect Dis 18(1):497
Nordgren J, Bonkoungou IJ, Nitiema LW, Sharma S, Ouermi D, Simpore J et al (2012) Rotavirus in diarrheal children in rural Burkina Faso: high prevalence of genotype G6P[6]. Infect Genet Evol 12(8):1892–1898
Cubitt WD, Steele AD, Iturriza M (2000) Characterisation of rotaviruses from children treated at a London hospital during 1996: emergence of strains G9P2A[6] and G3P2A[6]. J Med Virol 61(1):150–154
Wahyuni RM, Utsumi T, Dinana Z, Yamani LN, Juniastuti WIS et al (2021) Prevalence and distribution of rotavirus genotypes among children with acute gastroenteritis in areas other than Java Island, Indonesia, 2016–2018. Front Microbiol 12:672837
Dong S, Huang D, Wang Z, Zhang G, Zhang F, Sai L (2021) Clinical and molecular epidemiological characterization of rotavirus infections in children under five years old in Shandong province, China. Arch Virol 166(9):2479–2486
Degiuseppe JI, Stupka JA, Argentinean Rotavirus Surveillance Network (2021) Emergence of unusual rotavirus G9P[4] and G8P[8] strains during post vaccination surveillance in Argentina, 2017–2018. Infect Genet Evol 93:104940
Clark HF, Hoshino Y, Bell LM, Groff J, Hess G, Bachman P et al (1987) Rotavirus isolate WI61 representing a presumptive new human serotype. J Clin Microbiol 25:1757–1762
Wu FT, Bányai K, Jiang B, Liu LT, Marton S, Huang YC et al (2017) Novel G9 rotavirus strains co-circulate in children and pigs, Taiwan. Sci Rep 7:40731
Bozdayi G, Altay A, Yahiro T, Ahmed S, Meral M, Dogan B et al (2016) Re-emergence of genotype G9 during a five-and-a-half-year period in Turkish children with rotavirus diarrhea. Arch Virol 161(10):2879–2884
Sashina TA, Morozova OV, Epifanova NV, Novikova NA (2017) Predominance of new G9P[8] rotaviruses closely related to Turkish strains in Nizhny Novgorod (Russia). Arch Virol 162(8):2387–2392
Kaplon J, Grangier N, Pillet S, Minoui-Tran A, Vabret A, Wilhelm N et al (2018) Predominance of G9P[8] rotavirus strains throughout France, 2014–2017. Clin Microbiol Infect 24(6):660.e1-660.e4
Zuridah H, Kirkwood CD, Bishop RF, Bogdanovic-Sakran N, Yap KL (2009) Molecular characterization and epidemiology of rotavirus isolates obtained from children with diarrhoea in Malaysia. Med J Malaysia 64(3):193–196
Hung LC, Wong SL, Chan LG, Rosli R, Ng AN, Bresee JS (2006) Epidemiology and strain characterization of rotavirus diarrhea in Malaysia. Int J Infect Dis 10(6):470–474
Sum MS, Perera D, Ramji N, Elie F, Cardosa MJ (2008) Molecular characterization of rotavirus A associated with outbreaks of acute gastroenteritis in Sarawak in 2001 and 2007. Int J Infect Dis 12(1):e91–e92
Phan TG, Okitsu S, Maneekarn N, Ushijima H (2007) Genetic heterogeneity, evolution and recombination in emerging G9 rotaviruses. Infect Genet Evol 7(5):656–663
Fujii Y, Oda M, Somura Y, Shinkai T (2020) Molecular characteristics of novel mono-reassortant G9P[8] rotavirus A strains possessing the NSP4 gene of the E2 genotype detected in Tokyo, Japan. Jpn J Infect Dis 73(1):26–35
Hoa-Tran TN, Nakagomi T, Vu HM, Nguyen TTT, Takemura T, Hasebe F et al (2020) Detection of three independently-generated DS-1-like G9P[8] reassortant rotavirus A strains during the G9P[8] dominance in Vietnam, 2016–2018. Infect Genet Evol 80:104194
Zhou X, Wang YH, Pang BB, Chen N, Kobayashi N (2020) Surveillance of human rotavirus in Wuhan, China (2011–2019): predominance of G9P[8] and emergence of G12. Pathogens 9(10):810
Teodoroff TA, Tsunemitsu H, Okamoto K, Katsuda K, Kohmoto M, Kawashima K et al (2005) Predominance of porcine rotavirus G9 in Japanese piglets with diarrhea: close relationship of their VP7 genes with those of recent human G9 strains. J Clin Microbiol 43(3):1377–1384
Gupta S, Tiku VR, Gauhar M, Khatoon K, Ray P (2021) Genetic diversity of G9 rotavirus strains circulating among diarrheic children in North India: a comparison with 116E rotavirus vaccine strain. Vaccine 39:646–651
Dyall-Smith ML, Lazdins I, Tregear GW, Holmes IH (1986) Location of the major antigenic sites involved in rotavirus serotype-specific neutralization. Proc Natl Acad Sci USA 83(10):3465–3468
Burke RM, Tate JE, Kirkwood CD, Steele AD, Parashar UD (2019) Current and new rotavirus vaccines. Curr Opin Infect Dis 32(5):435–444
Patel M, Shane AL, Parashar UD, Jiang B, Gentsch JR, Glass RI (2009) Oral rotavirus vaccines: how well will they work where they are needed most? J Infect Dis 200 Suppl 1(01):S39-48
Fukuda Y, Tsugawa T, Nagaoka Y, Ishii A, Nawa T, Togashi A et al (2021) Surveillance in hospitalized children with infectious diseases in Japan: pre- and post-coronavirus disease 2019. J Infect Chemother 27(11):1639–1647
Ullrich A, Schranz M, Rexroth U, Hamouda O, Schaade L, Diercke M et al (2021) Impact of the COVID-19 pandemic and associated non-pharmaceutical interventions on other notifiable infectious diseases in Germany: an analysis of national surveillance data during week 1–2016—week 32–2020. Lancet Reg Health Eur 6:100103
Ahn SY, Park JY, Lim IS, Chae SA, Yun SW, Lee NM et al (2021) Changes in the occurrence of gastrointestinal infections after COVID-19 in Korea. J Korean Med Sci 36(24):e180
Acknowledgements
The authors would like to thank the Director General of Health Malaysia for permission to publish this article.
Funding
This study was funded by the Ministry of High Education under the Fundamental Research Grant Scheme (FRGS/1/2017/SKK11/UMS/01/2) (URL: https://mygrants.gov.my) to KA.
Author information
Authors and Affiliations
Contributions
KA: conceptualized the study, analyzed data, acquired funds, performed project administration, supervised, reviewed, edited, and wrote the final manuscript. LNA wrote the original draft, curated data, analyzed data, performed investigations, performed laboratory procedures, reviewed, edited, and wrote the final manuscript. JLJ curated data, analyzed data, performed investigations, performed laboratory procedures, reviewed, edited, and wrote the final manuscript. DM analyzed data, performed investigations, performed laboratory procedures, supervised, reviewed, edited, and wrote the final manuscript. AZC analyzed data, performed investigations, performed project administration, reviewed, edited, and wrote the final manuscript. AKM analyzed data, performed investigations, performed project administration, reviewed, edited, and wrote the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Handling Editor: Tim Skern.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Amit, L.N., John, J.L., Mori, D. et al. Increase in rotavirus prevalence with the emergence of genotype G9P[8] in replacement of genotype G12P[6] in Sabah, Malaysia. Arch Virol 168, 173 (2023). https://doi.org/10.1007/s00705-023-05803-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05803-9