
Abstract
Here, we report the discovery and complete genome sequence of a novel virus, designated as “Phytophthora heveae alphaendornavirus 1” (PhAEV1), from a single isolate of the plant pathogenic oomycete Phytophthora heveae (kingdom Stramenipila) isolated from a tropical evergreen lowland rainforest in northern Vietnam. PhAEV1 was detected by both cellulose affinity chromatography of dsRNA and high-throughput sequencing of total RNA, and its presence and sequence were confirmed by RT-PCR and Sanger sequencing. The PhAEV1 genome, 12,820 nucleotides (nt) in length, was predicted to encode a single large polyprotein with the catalytic core domain of viral (superfamily 1) RNA helicase (HEL, amino acid [aa] positions 1,287-1,531), glycosyltransferase (GT, aa positions ca. 2,800-3,125), and RNA-directed RNA polymerase (RdRp, aa positions 3,875-4,112). PhAEV1 is the most similar to Phytophthora cactorum alphaendornavirus 3, sharing 39.4% and 39.1% nt and aa sequence identity, respectively. In addition to the first 5′-terminal AUG codon, three additional in-frame methionine codons were found in close proximity (nt 14-16, 96-98, and 176-178), suggesting potential additional translation initiation sites. Conserved RdRp motifs (A-E) similar to those detected in related endornaviruses were identified in PhAEV1, as well as in several previously described alphaendornaviruses from other Phytophthora species in which these motifs had not been identified previously. Phylogenetic analysis showed that PhAEV1 clusters with members of the genus Alphaendornavirus in the family Endornaviridae and is basal to two other alphaendornaviruses described from another oomycete, Phytophthora cactorum. PhAEV1 is the first virus reported in P. heveae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Phytophthora heveae (A.W. Thomps.) is a plant-pathogenic homothallic oomycete (Stramenipila (syn. Heterokonta), Oomycota, Peronosporales, Peronosporaceae) that is placed taxonomically in phylogenetic major clade 5 of the genus Phytophthora. It is known as a root and bark pathogen of Theobroma L., Persea Mill. and Rhododendron L., distributed across East and Southeast Asia; Central, North, and South America; and Oceania [1,2,3,4,5,6,7]. The genus Phytophthora is entirely composed of plant pathogens and includes some of the most devastating species threatening agricultural, forest, and natural ecosystems worldwide [3, 8]. Mycoviruses, which, by broad definition are fungal viruses and oomycete viruses, generally infect their hosts persistently, in most cases causing cryptic infections with no apparent symptoms. However, they have attracted considerable attention because of their potential to reduce fungal virulence and cause “hypovirulence” [9]. Previously, there have been no records of viral infection in P. heveae, although a range of RNA viruses from different families with diverse genome organizations have been described in other Phytophthora species [10,11,12,13,14,15,16,17]. Additionally, several ORFan sequences of putative viral origin have been detected in Phytophthora castaneae Katsura & K. Uchida [10], which belongs to the same phylogenetic clade (clade 5) as P. heveae [6]. The family Endornaviridae includes linear positive (+) sense, single-stranded (ss) RNA viruses that do not form true virions. Although previously classified as double-stranded (ds) RNA viruses, their dsRNA form is now considered a replicative intermediate that accumulates during replication of the ssRNA genome [18]. Their genome contains a single large open reading frame (ORF) encoding a polyprotein ranging in size from 3,217 to 5,825 amino acids (aa) with a highly conserved RNA-directed RNA polymerase (RdRp) domain and an RNA helicase domain located in the C-terminal and middle region, respectively. Some endornaviruses encode other functional domains, including methyltransferase, glycosyl transferase, cysteine-rich, phytoreo_S7, and capsular polysaccharide synthase domains [18, 19]. The family is divided into two genera: Alphaendornavirus, which includes viruses that infect plants, fungi, and oomycetes [19], and Betaendornavirus, which includes viruses that infect ascomycetous fungi [20]. A characteristic that is common to many alphaendornaviruses is the occurrence of a site-specific break (nick) located at a 5’-terminal end of the coding strand [18], the biological significance of which is unknown. Alphaendornaviruses have been reported in several members of the genus Phytophthora, including the Phytophthora taxon “douglas-fir” [21], P. ramorum Werres, De Cock & Man in ’t Veld [22], P. cactorum [13], a Vietnamese isolate of P. castaneae [10], and Phytophthora spp. infecting asparagus in Japan [15]. Compared to viruses of ascomycetes or basidiomycetes, viruses infecting oomycetes, including members of the genus Phytophthora, have not been extensively studied, and overall, our knowledge of the effects of viruses on their Phytophthora hosts is limited [23]. Plant and fungal endornaviruses are usually associated with asymptomatic infections, and only a few viruses are associated with phenotypic alterations. Vicia faba endornavirus VfEV is associated with cytoplasmatic male sterility traits in the broad bean Vicia faba L., while Helicobasidium mompa endornavirus 1 (HmEV1) infecting a basidiomycete Helicobasidium mompa Nobuj. Tanaka 1891, appears to confer hypovirulence to its host [19]. Infection by Rhizoctonia solani endornavirus 1 (RsEV1), the putative member of the family Endornaviridae with the largest genome reported to date, causes metabolic changes in its host Rhizoctonia solani Kühn, resulting in hypovirulence [24]. Additionally, Japanese isolates of Phytophthora sp. coinfected with two endornaviruses have been reported to exhibit slower growth and reduced hyphal density while producing abundant zoosporangia. Endornaviruses can also alter the fungicide sensitivity of their oomycete host [15].
Here, we report the discovery of a novel alphaendornavirus, the first virus to be described in the oomycetous plant pathogen P. heveae.
Provenance of the virus material
P. heveae isolate VN787 was originally isolated in the spring of 2016 from a mixed rhizosphere soil sample of two native Saraca dives Pierre, Fl. Forest. Cochinch. t. 386 B. (1898) (Fabaceae) trees growing in a tropical evergreen lowland rainforest in Cuc-Phuong National Park in northern Vietnam (Supplementary Table S1). The isolate was obtained from the culture collection of T. Jung hosted at the Phytophthora Research Centre (PRC), Mendel University in Brno, and was identified based on molecular sequences of the internal transcribed spacer (ITS1-5.8S-ITS2) region of the ribosomal DNA and the cytochrome c oxidase subunit 1 mitochondrial gene (cox1), and also based on microscopy of its morphological features [2]. VN787 was maintained on V8-juice agar (V8A; 100 ml of V8 juice [Hermann Pfanner Getränke, Lauterach, Austria], 16 g of agar [Sigma-Aldrich, St. Louis, MO, USA], 2 g of CaCO3, and 900 ml of distilled water per liter) at 20-23 °C in the dark. For extractions of total RNA and dsRNA, mycelia were grown on orange serum agar (OSA) (HiMedia Laboratories, Kennett Square, PA, USA) covered with a cellophane membrane (EJA08-100; Gel Company Inc., San Francisco, CA, United States). dsRNA was isolated from approximately 150 mg of mycelia using a method based on cellulose affinity chromatography as described by Suzuki et al. [25] and visualized in a 1% agarose gel after electrophoresis at 50 V for 60 min and staining with ethidium bromide. Total RNA extraction and cDNA synthesis were performed using RNAzol® RT (Sigma-Aldrich, Steinheim, Germany) following the manufacturer’s recommendations with some modifications, and a High-Capacity cDNA Reverse Transcription Kit (Applied Biosciences, Park Ave, NY, USA), respectively, and the quality and/or quantity of the preparations were estimated as described previously [10]. The total RNA of VN787 was sent to SEQme s.r.o (Dobris, Czech Republic) for RNA library construction and high-throughput sequencing (HTS) in a pool with eight other P. heveae isolates from Vietnam (Supplementary Table S1), whose total RNA was prepared as described above. Ribosomal RNA (rRNA) was depleted using an NEBNext rRNA Depletion Kit (Human/Mouse/Rat) (New England Biolabs (NEB) Ipswich, MA, USA), and the library was constructed using an NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB). The library was sequenced in paired-end (2 × 150 base-pairs [bp]) mode on an Illumina NovaSeq6000 (DS-150) platform using a NovaSeq S4 v1.5 reagent kit, together with a control library PhiX Control Kit v3 (Illumina). The quality was inspected using the FastQC-0.10.1 program [26] and the reads were assembled de novo using Trinity v2.6.5 [27]. The Trinity contigs were compared to a custom virus protein database using the BLASTx (BLASTX 2.10.0+) algorithm [28] with the E-value set to 10−5. All sequences showing significant similarity to known viruses were aligned in BLASTn (NCBI BLAST+ 2.12.0) to eliminate contigs potentially originating from the host or contaminants. Finally, contigs of interest were imported into Geneious Prime® 2023.0.4 (Biomatters, Auckland, New Zealand) for further manipulation, including further assembly using Geneious de novo assembler, specifying medium-low sensitivity. Virus-specific primers used for virus detection, identification of the ends of viral sequences, and confirmation of internal regions where the contigs overlapped or showed signs of variability were designed using Primer 3 2.3.7 in Geneious Prime® 2020.2.3 (Supplementary Table S2). The depth of coverage was calculated using Bowtie 2 v2.3.0 [29] as described by Raco et al. [10]. The 5′- and 3′-terminal sequences of the dsRNA segment were determined by performing RNA-ligase-mediated rapid amplification of cDNA ends (RLM-RACE) [25] and cloning into pGEM®-T Easy Vector System (Promega, Madison, WI, USA), followed by sequencing of the cloned inserts in both directions by the Sanger method. The NCBI CD-search tool [30] and HHpred server on the MPI Bioinformatics Toolkit website [31] were used to conduct a search for putative conserved domains (both tools last accessed on the 15th of April 2023). Phylogenetic analysis was performed based on RdRP amino acid (aa) and viral helicase domain sequences. Sequences were aligned using MAFFT V7.450 [32] in Geneious Prime® 2023.0.4, and TrimAI v. 1.3 software on the Phylemon 2.0 online platform (http://phylemon2.bioinfo.cipf.es) was used to remove unreliable alignment regions. Bayesian phylogenetic trees were constructed using MrBayes 3.2.6 [33], implemented in Geneious Prime® 2023.0.4.
Sequence properties
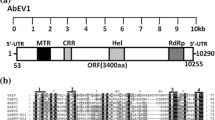
Total RNA sequencing generated 509,549,928 reads in total and produced 52,089 Trinity-assembled contigs. BLAST analysis confirmed the presence of multiple virus-like contigs resembling members of several virus families (data not shown), including putative members of the family Endornaviridae. The virus characterized here, designated as "Phytophthora heveae alphaendornavirus 1" (PhAEV1), was assembled by Geneious assembler from 20 Trinity contigs ranging in size from 238 bp to 5,262 bp (final contig length obtained by assembly, 12,778 bp, with partial terminal sequences undetermined). We confirmed by dsRNA (Fig. 1A) and RT-PCR analysis (Fig. 1B) that PhAEV1 was present in a single isolate, VN787, but not in the other oomycetous strains used for HTS (Fig. 1B). Virus-specific primers were used to validate regions of overlapping contigs (Fig. 1C), and primer set PhAEV_1_set_1a (Supplementary Table S2) was used for virus detection by RT-PCR in other available cDNAs of the sequencing pool (Fig. 1B). The full-length 12,820-bp-long genomic sequence of PhAEV1, including the 5′-, and 3′-terminal sequences determined by RLM-RACE, was deposited in the GenBank database under accession number OQ786863. Out of the total number of reads, 27,923 mapped to the PhAEV1 sequence. The estimated average depth of coverage for PhAEV1 was 326.71. The 5′-terminal sequence was determined based on 13 individual clones (Fig. 1D). The first nucleotide differed among the clones; five clones had G, four had C, two had T, and two were missing an additional residue. The 3’-terminal sequence was determined based on 10 individual clones that were all identical (Fig. 1D). A single large open reading frame (ORF) of 12,753 nt (4,250 aa) encoding a single polyprotein was detected on the positive (+) strand (frame 2), starting at nt 5 and ending at nt 12,757 (calculated molecular weight, 479.615 kDa) (Fig. 1E). The first methionine codon was observed at nt positions 5-8, but three other in-frame methionine codons were detected in close proximity, 6, 88, and 168, nucleotides downstream from the first AUG, suggesting that these could represent additional translation initiation sites (Fig. 1E). Between the second and third AUG codon, an unusual stretch of five Cs, five Gs, and an A-rich region with 20 A residues was detected. Similar elements have also been observed in Phytophthora cactorum alphaendornavirus 3 (PcAEV3). According to Kozak [34, 35] the optimal sequence for initiation by ribosomal subunits has either a purine base at position nt −3, or a G in position nt +4, or both. Therefore, the second methionine codon with A (-3) and G (+4) occurs in an optimal context for translation initiation. Since it remains to be determined which AUG serves as the main initiation site, the first AUG triplet is tentatively regarded as an initiation codon for PhAEV1. The polyprotein of PhAEV1 encodes a catalytic core domain of RdRp common to members of the family Endornaviridae (accession cd23255) at nt 11,627-12,340 (E-value, 1.86e-96; CD length, 237 aa), and a viral (superfamily 1) RNA helicase (HEL) domain, which is essential for virus replication, at nt 3,863-4,597 (E-value, 6.70e-07; CD length, 227 aa). In addition, a glycosyltransferase (GT) domain was detected using the HHpred tool, but not NCBI CD-search, at the approximate aa position from ~2,800 to 3,125. Five conserved motifs (A-E) were identified by sequence similarity in the RdRp region of PhAEV1 and other related viruses (Fig. 1F). The conserved residues of motifs A-D were fairly similar to those reported previously by Hacker et al. [21]. Additionally, conserved motifs were analyzed in sequences from several other alphaendornaviruses reported previously from Phytophthora species for which conserved motifs had not been reported previously (Fig. 1F). According to BLASTx analysis, the nucleotide sequence of PhAEV1 is most similar to the polyprotein sequence of PcAEV3 (QUA12642.1) (query coverage, 95.0%; E-value, 0.0; percent identity, 39.4%). At the protein level, the similarity of these two sequences is 39.1% (query coverage, 98.0%; E-value, 0.0). PhAEV1 also shows similarity to Krauss’ spikemoss associated endorna-like virus (accession no. CAH2618724.1; query coverage, 87.0%; E-value, 0.0; percent identity, 28.4%), and Diatom colony associated dsRNA virus 15 (accession no. YP_009552081.1; query coverage, 90.0%; E value, 0.0; percent identity, 27.1%) at the nucleotide level. Phylogenetic analysis showed that PhAEV1 forms a cluster with its descendants, Phytophthora cactorum alphaendornavirus 1 and 2 (PcAEV1 and PcAEV2) (Fig. 2A and B). According to the species demarcation criteria for the genus Alphaendornavirus established by the International Committee on Taxonomy of Viruses (ICTV), for viruses to be considered members of different species, they should have an overall nucleotide sequence identity below 75.0%, and their genomes should be greater than 1.9 kb in size [18]. Its phylogeny, genome organization, and sequence divergence support the classification of PhAEV1 as a member of a new species in the genus Alphaendornavirus, family Endornaviridae.

(A) dsRNA profile of P. heveae isolate VN787. The PhAEV1 replicative form was visualized by ethidium bromide staining in a 1% agarose gel. M1, GeneRuler 1 kb DNA Ladder; M2, dsRNA virus marker consisting of the genomic dsRNA segments or replicative form of Cryphonectria hypovirus 1 (CHV1), yado-nushi virus 1 (YnV1), and yado-kari virus 1 (YkV1), [36, 37]. (B) RT-PCR detection of PhAEV1 using the virus-specific primer set PhAEV1_set_1a. Seven out of nine oomycetous isolates used in this study for high-throughput sequencing were tested. Two isolates (VN529 and VN789), whose RNA was pooled for HTS, lost viability during storage. (C) Amplification of the PhAEV1 internal sequences by RT-PCR. Virus-specific primer sets (1, PhAEV1_ set_1a; 2, PhAEV1_ set_2; 3, PhAEV1_ set_2a; 4, PhAEV1_ set_3; 5, PhAEV1_ set_3a; 6, PhAEV1_ set_4; 7, PhAEV1_ set_4a; 8, PhAEV1_ set_5; 9, PhAEV1_ set_6) are listed in Supplementary Table S2. (D) Amplification of the 5′- and 3′- terminal sequences of the PhAEV1 genomic RNA. RLM-RACE was performed on PhAEV1 dsRNA. Amplified cDNA fragments were analyzed by electrophoresis in a 2% agarose gel. M, 100 bp Plus DNA Ladder (15628019, Thermo Fisher Scientific). (E) Schematic representation of the genome structure of PhAEV1. (F) Multiple aa sequence alignment of the conserved RdRp region of PhAEV1 and related viruses, performed using MAFFT v7.450 (BLOSUM45 scoring matrix) in Geneious Prime® 2023.0.4. Gray boxes at the bottom of the alignment with letters A-E indicate conserved motifs. Conserved residues are indicated by green boxes and highlighted with a black background, while semi-conserved residues are highlighted in gray. Conserved residues characteristic of alphaendornaviruses that infect Phytophthora spp. are indicated by yellow boxes with a star symbol. Numbers with a grey background at the bottom of the alignment indicate the number of residues that were deleted for better visualization. Virus abbreviations: BaEV, Basella alba endornavirus (NC043109); PaEV, Persea americana endornavirus (YP005086952); YmEV, Yerba mate alphaendornavirus (YP009046830); HvEV, Hordeum vulgare alphaendornavirus (YP009212849); PvEV1, Phaseolus vulgaris endornavirus 1 (YP009506353); RcEV1, Rhizoctonia cerealis endornavirus 1 (YP008719905); WBEV1, winged bean alphaendornavirus 1 (YP009305414); HPEV, hot pepper alphaendornavirus (YP009165596); BPEV, bell pepper alphaendornavirus (YP004765011); OrEV, Oryza rufipogon alphaendornavirus (YP438202); OsEV, Oryza sativa alphaendornavirus (YP438200); GEEV, grapevine endophyte alphaendornavirus (YP007003829); EcEV, Erysiphe cichoracearum alphaendornavirus (YP009225663); PEV1, Phytophthora alphaendornavirus 1 (YP241110); PEV2, Phytophthora endornavirus 2 (BCL84886); PEV3, Phytophthora endornavirus 3 (BCL84887); PcAEV1, Phytophthora cactorum alphaendornavirus 1 (QUA12640); PcAEV2, Phytophthora cactorum alphaendornavirus 2 (QUA12641); PcAEV3, Phytophthora cactorum alphaendornavirus 3 (QUA12642); PcaRV5, Phytophthora castaneae RNA virus 5 (USL98317); PhAEV1, Phytophthora heveae alphaendornavirus 1 (OQ786863)

Bayesian trees showing phylogenetic relationships of PhAEV1. The trees were constructed using the aa sequences of the (A) RdRp and (B) viral helicase domains of PhAEV1 (indicated by a purple star) and those of other endornaviruses. Multiple sequence alignments performed using MAFFT v7.450 (BLOSUM45 scoring matrix) consisted of 201 (A) and 175 (B) aa sites. The analyses were conducted using MrBayes 3.2.6., including grapevine leafroll-associated virus 1 (genus Ampelovirus, family Closteroviridae) as an outgroup. Percentage posterior probabilities are displayed at branch nodes. The scale bar shows 0.3 (A) and 0.5 (B) aa substitutions per site.
Sequence data availability
The complete genomic nucleotide sequence of Phytophthora heveae alphaendornavirus 1 (PhAEV1) has been deposited in the NCBI GenBank database under accession number OQ786863. The dataset generated by HTS of total RNA contains unpublished viral sequences and is therefore available from the corresponding author only upon reasonable request.
References
Jung T, Chang TT, Bakonyi J et al (2017) Diversity of Phytophthora species in natural ecosystems of Taiwan and association with disease symptoms. Plant Pathol 66:194–211. https://doi.org/10.1111/ppa.12564
Jung T, Scanu B, Brasier CM et al (2020) A survey in natural forest ecosystems of Vietnam reveals high diversity of both new and described Phytophthora taxa including P. ramorum. Forests 11:93. https://doi.org/10.3390/f11010093
Erwin CD, Ribeiro KO (1996) Phytophthora diseases worldwide. The American Phytopathological Society, St. Paul
Burgess TI, White D, McDougall KM et al (2017) Distribution and diversity of Phytophthora across Australia. Pac Conserv Biol 23:150–162. https://doi.org/10.1071/PC16032
Legeay J, Husson C, Boudier B et al (2020) Surprising low diversity of the plant pathogen Phytophthora in Amazonian forests. Environ Microbiol 22:5019–5032. https://doi.org/10.1111/1462-2920.15099
Weir BS, Paderes EP, Anand N et al (2015) A taxonomic revision of Phytophthora clade 5 including two new species, Phytophthora agathidicida and P. cocois. Phytotaxa 205:21–38. https://doi.org/10.11646/phytotaxa.205.1.2
Zeng HC, Ho HH, Zheng FC (2009) A survey of Phytophthora species on Hainan Island of South China. J Phytopathol 157:33–39. https://doi.org/10.1111/J.1439-0434.2008.01441.X
Brasier C, Scanu B, Cooke D, Jung T (2022) Phytophthora: an ancient, historic, biologically and structurally cohesive and evolutionarily successful generic concept in need of preservation. IMA Fungus. https://doi.org/10.1186/s43008-022-00097-z
Ghabrial SA, Suzuki N (2009) Viruses of plant pathogenic fungi. Annu Rev Phytopathol 47:353–384. https://doi.org/10.1146/annurev-phyto-080508-081932
Raco M, Vainio EJ, Sutela S et al (2022) High diversity of novel viruses in the tree pathogen Phytophthora castaneae revealed by high-throughput sequencing of total and small RNA. Front Microbiol 13:1703. https://doi.org/10.3389/fmicb.2022.911474
Botella L, Jung T (2021) Multiple viral infections detected in Phytophthora condilina by total and small RNA sequencing. Viruses 13:620. https://doi.org/10.3390/v13040620
Botella L, Jung MH, Rost M, Jung T (2022) Natural populations from the Phytophthora palustris complex show a high diversity and abundance of ssRNA and dsRNA viruses. J Fungi 8(11):1118. https://doi.org/10.3390/JOF8111118
Poimala A, Parikka P, Hantula J, Vainio EJ (2021) Viral diversity in Phytophthora cactorum population infecting strawberry. Environ Microbiol 23:5200–5221. https://doi.org/10.1111/1462-2920.15519
Poimala A, Vainio EJ (2020) Complete genome sequence of a novel toti-like virus from the plant-pathogenic oomycete Phytophthora cactorum. Arch Virol 165:1679–1682. https://doi.org/10.1007/s00705-020-04642-2
Uchida K, Sakuta K, Ito A et al (2021) Two novel endorna viruses co-infecting a Phytophthora pathogen of Asparagus officinalis modulate the developmental stages and fungicide sensitivities of the host oomycete. Front Microbiol. https://doi.org/10.3389/fmicb.2021.633502
Cai G, Hillman BI (2013) Phytophthora viruses. In: Advances in virus research, vol 86. Academic Press Inc., pp 327–350
Xu Z, Khalifa ME, Frampton RA et al (2022) Characterization of a novel double-stranded RNA virus from Phytophthora pluvialis in New Zealand. Viruses. https://doi.org/10.3390/v14020247
Valverde RA, Khalifa ME, Okada R et al (2019) ICTV virus taxonomy profile: Endornaviridae. J Gen Virol 100:1024–1025. https://doi.org/10.1099/jgv.0.001277
Fukuhara T, Gibbs MJ (2012) Family Endornaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego, pp 519–521
Khalifa ME, Pearson MN (2014) Molecular characterisation of an endornavirus infecting the phytopathogen Sclerotinia sclerotiorum. Virus Res 189:303–309. https://doi.org/10.1016/J.VIRUSRES.2014.06.010
Hacker CV, Brasier CM, Buck KW (2005) A double-stranded RNA from a Phytophthora species is related to the plant endornaviruses and contains a putative UDP glycosyltransferase gene. J Gen Virol 86(5):1561–1570. https://doi.org/10.1099/vir.0.80808-0
Kozlakidis Z, Brown NA, Jamal A et al (2010) Incidence of endornaviruses in Phytophthora taxon douglasfir and Phytophthora ramorum. Virus Genes 40:130–134. https://doi.org/10.1007/s11262-009-0421-7
Poimala A, Raco M, Haikonen T et al (2022) Bunyaviruses affect growth, sporulation, and elicitin production in Phytophthora cactorum. Viruses 14(12):2596. https://doi.org/10.3390/v14122596
Zheng L, Shu C, Zhang M et al (2019) Molecular characterization of a novel endornavirus conferring hypovirulence in rice sheath blight fungus Rhizoctonia solani AG-1 IA strain GD-2. Viruses 11(2):178. https://doi.org/10.3390/v11020178
Suzuki N, Supyani S, Maruyama K, Hillman BI (2004) Complete genome sequence of Mycoreovirus-1/Cp9B21, a member of a novel genus within the family Reoviridae, isolated from the chestnut blight fungus Cryphonectria parasitica. J Gen Virol 85:3437–3448. https://doi.org/10.1099/VIR.0.80293-0
Andrews S (2010) Babraham bioinformatics—FastQC a quality control tool for high throughput sequence data. In: Soil. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 5 Apr 2023
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. https://doi.org/10.1038/nbt.1883
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402. https://doi.org/10.1093/nar/25.17.3389
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. https://doi.org/10.1038/nmeth.1923
Marchler-Bauer A, Bryant SH (2004) CD-Search: protein domain annotations on the fly. Nucleic Acids Res 32:327–331. https://doi.org/10.1093/nar/gkh454
Zimmermann L et al (2018) A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol 430(15):2237–2243
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17(8):754–755. https://doi.org/10.1093/bioinformatics/17.8.754
Kozak M (1981) Possible role of flanking nucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes. Nucleic Acids Res 9:5233–5252. https://doi.org/10.1093/NAR/9.20.5233
Kozak M (1986) Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44(2):283–292. https://doi.org/10.1016/0092-8674(86)90762-2
Shapira R, Choi GH, Nuss DL (1991) Virus-like genetic organization and expression strategy for a double-stranded RNA genetic element associated with biological control of chestnut blight. EMBO J 10:731-739. https://doi.org/10.1002/j.1460-2075.1991.tb08004.x
Zhang R, Hisano S, Tani A, et al (2016) A capsidless ssRNA virus hosted by an unrelated dsRNA virus. Nat Microbiol 1:15001 https://doi.org/10.1038/nmicrobiol.2015.1
Acknowledgements
MR is thankful to Hideki Kondo (Institute of Plant Science and Resources, Okayama University) for valuable advice related to phylogenetic analysis, and to Sakae Hisano and Kazuyuki Maruyama (both Institute of Plant Science and Resources, Okayama University), for technical support. Computational resources were provided by the e-INFRA CZ project (ID:90140), supported by the Ministry of Education, Youth, and Sports of the Czech Republic.
Funding
Open access publishing supported by the National Technical Library in Prague. This research was supported by the Specific University Research Fund of the Faculty of Forestry and Wood Technology, Mendel University in Brno LDF_VP_2021047, the European Regional Development Fund, project “Phytophthora Research Centre,” Reg. No. CZ.02.1.01/0.0/0.0/15_003/0000453, and Grants-in-Aid for Scientific Research (S), Research on Innovative Areas, and Grants-in-Aid for JSPS from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (KAKENHI 21H05035, 21K18222, 16H06436, 16H06429, and 16K21723 to NS).
Author information
Authors and Affiliations
Contributions
Original idea: MR, TJ, LB. Conceptualization: MR, NS. Investigation and formal analysis: MR. Writing—original draft preparation: MR. Writing—review and editing: NS, TJ. Providing Phytophthora isolates: TJ, MHJ, NMC. Funding acquisition: MR, TJ, LB, NS. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Ioly Kotta-Loizou.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raco, M., Jung, T., Horta Jung, M. et al. Sequence and phylogenetic analysis of a novel alphaendornavirus, the first virus described from the oomycete plant pathogen Phytophthora heveae. Arch Virol 168, 158 (2023). https://doi.org/10.1007/s00705-023-05786-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05786-7