
Abstract
Throughout East Asia, Europe, and North America, mammalian orthoreovirus (MRV), for which bats have been proposed to be natural reservoirs, has been detected in a variety of domestic and wild mammals, as well as in humans. Here, we isolated a novel MRV strain (designated as Kj22-33) from a fecal sample from Vespertilio sinensis bats in Japan. Strain Kj22-33 has a 10-segmented genome with a total length of 23,580 base pairs. Phylogenetic analysis indicated that Kj22-33 is a serotype 2 strain, the segmented genome of which has undergone reassortment with that of other MRV strains.
Similar content being viewed by others
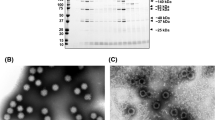

Avoid common mistakes on your manuscript.
Reoviruses, members of the order Reovirales, are classified into two families, namely, Sedoreoviridae and Spinareoviridae, each of which comprises several genera. Organisms within a diverse spectrum of taxonomic groups, including protists, plants, fish, amphibians, birds, and mammals, serve as hosts for viruses from both of these families. Mammalian orthoreovirus is one of the 10 species comprising the genus Orthoreovirus within the family Spinareoviridae, and it can be further divided into four major serotypes (MRV1–4) based on antigenicity in neutralization and hemagglutination inhibition tests [1, 2].
Mammalian orthoreovirus (MRV) is a non-enveloped virus with a 10-segmented double-stranded RNA genome consisting of three large (L1–L3), three medium (M1–M3), and four small (S1–S4) segments. The L1, L2, L3, M1, M2, S1, S2, and S4 segments encode eight structural proteins (λ1, λ2, λ3, µ1, µ2, σ1, σ2, and σ3, respectively), whereas the M3 segment encodes two non-structural proteins (µNS and µNSC), and the S1 and S3 segments each encode a non-structural protein (σ1s and σNS, respectively). The external capsid protein σ1 plays a role in cell attachment and is the only determinant distinguishing the serotypes.
Since its initial isolation from the stools of children in 1954 [1], MRV has been reported in children with gastroenteritis [3,4,5]. In addition, strains of MRV have been detected continually in a wide range of mammals, including pigs [6], wild boars [7], cats [8], dogs [9], bats [10,11,12,13,14,15,16,17], and deer [18], and have also been found in wastewater [19], indicating the broad host range of this virus. Bats are particularly noteworthy in this regard, in that they are considered to be natural reservoirs of MRV, and a genetically diverse range of strains have been detected in different bat species [10]. However, although bat MRVs have been reported in multiple regions, including Europe [9, 11], the United States [13], China [10, 14,15,16], and Korea [17], to date, none of these viruses have been detected in Japanese bats, despite the presence of MRV in several mammals in Japan [6,7,8,9]. In this study, we detected and isolated a novel MRV strain from bats in Japan.
To determine whether bat MRVs are present in Japan, in July 2022, we collected fecal samples from Asian particolored bats (Vespertilio sinensis) in Saitama Prefecture, Japan. Suspensions of the collected feces were subsequently used to inoculate Vero cells expressing transmembrane serine protease 2 (Vero/TMPRSS2), which may support the replication of unknown viruses, including MRVs [20]. The inoculated cells were incubated at 37℃ in a 5% CO2 atmosphere, and at 3 days post-inoculation, we observed a clear cytopathic effect (CPE). The cell supernatant was passed through a 0.22-µm-pore-size filter, and the filtrate was used inoculate fresh Vero/TMPRSS2 cells. Consistently, we observed a CPE, thereby confirming the isolation of a virus (hereafter referred to as Kj22-33). To identify the viral genome type, we used two antiviral drugs – ribavirin and 5-iodo-2ʹ-deoxyuridine (IUDR) – which inhibit the growth of RNA and DNA viruses, respectively. Viral growth was found to be inhibited only in the presence of ribavirin, indicating that Kj22-33 has an RNA genome.
We purified the Kj22-33 isolate from the infected cell supernatant by ultracentrifugation with a 20% sucrose cushion and extracted the RNA using ISOGEN-LS (Nippon Gene, Toyama, Japan). An MGIEasy RNA Directional Library Prep Set (MGI, Shenzhen, China) was used to prepare a library for genome sequencing using a DNBSEQ G400RS high-throughput sequencer (MGI, Shenzhen, China). The dataset thus obtained was assembled de novo using CLC Genomics Workbench software (ver. 8; CLCbio, Aarhus, Denmark), and accordingly, we obtained sequences for 10 segments. The 5ʹ and 3ʹ regions of each segment were subsequently determined by RACE (rapid amplification of cDNA ends) using a SMARTer RACE 5'/3' Kit (Takara Bio, Shiga, Japan), on the basis of which we determined the full viral genome sequence (GenBank accession numbers LC752173–LC752182). NCBI BLAST searches revealed that all 10 segments showed high similarity (96.2% to 99.5% identity) to the genomic sequences of MRV strains isolated from different animal species (Table 1).
Phylogenetic analysis was performed based on sequences of each Kj22-33 genome segment (Fig. 1). Trees were constructed using the maximum-likelihood method based on the Tamura-Nei model in MEGA X [21]. The S1 sequence of Kj22-33 was found to group in the same clade as serotype 2 strains. However, although the S1 segment of Kj22-33 appears to be closely related to that of the strain MRV2/RpMRV-YN2012/bat/2012/China (YN2012), the other segments of Kj22-33 were observed to cluster in clades distant from that containing YN2012. MRV2/OV204/deer/2016/USA showed the highest nucleotide sequence similarity in the L3 and S2 genomic segments, but it also exhibited phylogenetic relatedness in the S2, S3, M3, L1, and L3 segments, suggesting an evolutionary association among these genomic segments. These findings suggest that Kj22-33 may have arisen as a consequence of genomic reassortment with the closely related YN2012 and OV204 strains, as well as others.

Phylogenetic analysis of the 10 genome segments of strain Kj22-33. Phylogenetic trees were constructed by the maximum-likelihood method with 1,000 bootstrap replicates using MEGA X software. The numbers at nodes denote bootstrap values based on 1000 replicates. The scale bar shows the evolutionary distance in terms of nucleotide substitutions per site. The virus strain Kj22-33 is shown in bold red. Virus strains are labeled as follows: MRV serotype/strain name/detection host or material/detection year/country/GenBank accession number. The four serotype groups are indicated on the right-hand side of the S1 segment tree.
In general, viruses exhibit a high degree of host specificity and are able to infect only a narrow range of hosts. In contrast, MRV has been established to have low host specificity and is accordingly characterized by a broad host range. In this study, we conducted a comprehensive phylogenetic analysis of all genomic segments and found that although segment S1 of the Kj22-33 isolate is closely related to the corresponding segment of the YN2012 strain obtained from Chinese bats, five out of the ten segments of Kj22-33 were most closely related to those of MRV identified in white-tailed deer in the USA, despite the large geographical distance. Given that the host of Kj22-33, the bat Vespertilio sinensis, is a common species in East Asia, and that it inhabits an environment quite distinct from that of the white-tailed deer, the reason for the apparently close relationship between these two geographically distant viruses remains unclear. Moreover, given its low host specificity, the global distribution and epidemiology of MRV remains largely undetermined, and further studies are therefore required to clarify its mode of spread.
Data availability
The complete genome sequences of the MRV strain identified in this study have been deposited in the GenBank database under the accession numbers LC752173–LC752182.
References
Sabin AB (1959) Reoviruses A new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described. Science 130:1387–1389. https://doi.org/10.1126/science.130.3386.1387
Attoui H, Biagini P, Stirling J, Mertens PP, Cantaloube JF, Meyer A, de Micco P, de Lamballerie X (2001) Sequence characterization of Ndelle virus genome segments 1, 5, 7, 8, and 10: evidence for reassignment to the genus Orthoreovirus, family Reoviridae. Biochem Biophys Res Commun 287:583–588. https://doi.org/10.1006/bbrc.2001.5612
Yamamoto SP, Motooka D, Egawa K, Kaida A, Hirai Y, Kubo H, Motomura K, Nakamura S, Iritani N (2020) Novel human reovirus isolated from children and its long-term circulation with reassortments. Sci Rep 120:963. https://doi.org/10.1038/s41598-020-58003-9
Giordano MO, Martínez LC, Isa MB, Ferreyra LJ, Canna F, Paván JV, Paez M, Notario R, Nates SV (2002) Twenty year study of the occurrence of reovirus infection in hospitalized children with acute gastroenteritis in Argentina. Pediatr Infect Dis J 21:880–882. https://doi.org/10.1097/00006454-200209000-00021
Rosa UA, Ribeiro GO, Villanova F, Luchs A, Milagres FAP, Komninakis SV, Tahmasebi R, Lobato MCABS, Brustulin R, Chagas RTD, Abrão MFNDS, Soares CVDA, Tinker RJ, Pandey RP, Raj VS, Sabino EC, Deng X, Delwart E, Costa ACD, Leal É (2019) First identification of mammalian orthoreovirus type 3 by gut virome analysis in diarrheic child in Brazil. Sci Rep 9:18599. https://doi.org/10.1038/s41598-019-55216-5
Hirahara T, Yasuhara H, Matsui O, Kodama K, Nakai M, Sasaki N (1988) Characteristics of reovirus type 1 from respiratory tract of pigs in Japan. Nihon Juigaku Zasshi 50:353–361. https://doi.org/10.1292/jvms1939.50.353
Zhang W, Kataoka M, Doan YH, Oi T, Furuya T, Oba M, Mizutani T, Oka T, Li TC, Nagai M (2021) Isolation and characterization of mammalian orthoreovirus type 3 from a fecal sample from a wild boar in Japan. Arch Virol 166:1671–1680. https://doi.org/10.1007/s00705-021-05053-7
Mochizuki M, Tamazumi T, Kawanishi A, Azuma T, Shimizu T (1992) Serotype 2 reoviruses from the feces of cats with and without diarrhea. J Vet Med Sci 54:963–968. https://doi.org/10.1292/jvms.54.963
Kokubu T, Takahashi T, Takamura K, Yasuda H, Hiramatsu K, Nakai M (1993) Isolation of reovirus type 3 from dogs with diarrhea. J Vet Med Sci 55:453–454. https://doi.org/10.1292/jvms.55.453
Li X, Sun X, Lu C, Kuang D, Han Y, Wang W, Tong P, Li N, Zhou J, Dai J (2020) Isolation and identification of two new strains of mammalian orthoreovirus from Chinese tree shrews. Arch Virol 165:1541–1550. https://doi.org/10.1007/s00705-020-04635-1
Lelli D, Moreno A, Lavazza A, Bresaola M, Canelli E, Boniotti MB, Cordioli P (2013) Identification of mammalian orthoreovirus type 3 in Italian bats. Zoonoses Public Health 60:84–92. https://doi.org/10.1111/zph.12001
Naglič T, Rihtarič D, Hostnik P, Toplak N, Koren S, Kuhar U, Jamnikar-Ciglenečki U, Kutnjak D, Steyer A (2018) Identification of novel reassortant mammalian orthoreoviruses from bats in Slovenia. BMC Vet Res 14:264. https://doi.org/10.1186/s12917-018-1585-y
Feng KH, Brown JD, Turner GG, Holmes EC, Allison AB (2022) Unrecognized diversity of mammalian orthoreovirus in North American bats. Virology 571:1–11. https://doi.org/10.1016/j.virol.2022.03.012
Yang XL, Tan B, Wang B, Li W, Wang N, Luo CM, Wang MN, Zhang W, Li B, Peng C, Ge XY, Zhang LB, Shi ZL (2015) Isolation and identification of bat viruses closely related to human, porcine and mink orthoreovirus. J Gen Virol 96:3525–3531. https://doi.org/10.1099/jgv.0.000314
Wang L, Fu S, Cao L, Lei W, Cao Y, Song J, Tang Q, Zhang H, Feng Y, Yang W, Liang G (2015) Isolation and identification of a natural reassortant mammalian orthoreovirus from least horseshoe bat in China. PLoS ONE 10(3):e0118598. https://doi.org/10.1371/journal.pone.0118598
Li Z, Liu D, Ran X, Liu C, Guo D, Hu X, Tian J, Zhang X, Shao Y, Liu S (2016) Characterization and pathogenicity of a novel mammalian orthoreovirus from wild short-nosed fruit bats. Infect Genet Evol 43:347–353. https://doi.org/10.1016/j.meegid.2016.05.039
Lo VT, Yoon SW, Noh JY, Jang SS, Na W, Song D, Jeong DG, Kim HK (2022) Characterization of replication and variations in genome segments of a bat reovirus, BatMRV/B19-02, by RNA-seq in infected Vero-E6 cells. Arch Virol 167:2133–2142. https://doi.org/10.1007/s00705-022-05534-3
Ahasan MS, Subramaniam K, Sayler KA, Loeb JC, Popov VL, Lednicky JA, Wisely SM, Campos Krauer JM, Waltzek TB (2019) Molecular characterization of a novel reassortment mammalian orthoreovirus type 2 isolated from a Florida white-tailed deer fawn. Virus Res 270:197642. https://doi.org/10.1016/j.virusres.2019.197642
Kitamura K, Takagi H, Oka T, Kataoka M, Ueki Y, Sakagami A (2021) Intertypic reassortment of mammalian orthoreovirus identified in wastewater in Japan. Sci Rep 11:12583. https://doi.org/10.1038/s41598-021-92019-z
Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, Tahara M, Kikuta H, Yanagi Y (2008) Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J Virol 82:8942–8946. https://doi.org/10.1128/JVI.00676-08
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Bio Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Funding
Open access funding provided by The University of Tokyo. This work was supported by the Japan Agency for Medical Research and Development (AMED) under grant numbers JP21fk0108602 and JP21fk0108615.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
This study did not involve experiments with human participants performed by any of the authors. Experiments with bat samples were approved by the Animal Experiment Committee of the Graduate School of Agricultural and Life Sciences at the University of Tokyo (approval number P21-058).
Additional information
Handling Editor: Zhenhai Chen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ichikawa, A., Katayama, M., Lai, H. et al. Isolation and genetic characterization of a mammalian orthoreovirus from Vespertilio sinensis in Japan. Arch Virol 168, 165 (2023). https://doi.org/10.1007/s00705-023-05782-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05782-x