
Abstract
In this study, the complete genome of a novel polyomavirus detected in a great cormorant (Phalacrocorax carbo) was characterized. The 5133-bp-long genome of the cormorant polyomavirus has a genomic structure typical of members of the genus Gammapolyomavirus, family Polyomaviridae, containing open reading frames encoding the large and small tumor antigens, viral proteins 1, 2, and 3, and the X protein. The large tumor antigen of the cormorant polyomavirus shares 45.6–50.4% amino acid sequence identity with the homologous sequences of other gammapolyomaviruses. These data, together with results of phylogenetic analysis, suggest that this cormorant polyomavirus should be considered the first member of a new species within the genus Gammapolyomavirus, for which we propose the name “Phalacrocorax carbo polyomavirus 1”.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Members of the family Polyomaviridae infect mammals, birds, and fish [1,2,3]. The biology of avian and mammalian polyomaviruses differs significantly. Avian polyomaviruses are not highly species-specific, and they cause acute, fulminant, and often fatal disease in their susceptible hosts. In contrast, mammalian polyomaviruses show rigorous host species specificity and typically cause inapparent infections in immunocompetent hosts [2]. The family Polyomaviridae comprises eight genera. Viruses of the genera Alpha-, Beta-, and Deltapolyomavirus, as well as the recently established genera Epsilon- and Zetapolyomavirus have been detected from mammals, whereas members of the genus Gammapolyomavirus infect birds. Polyomaviruses of fish have been assigned to the new genera Etapolyomavirus and Thetapolyomavirus [1].
Polyomaviruses are characterized by nonenveloped icosahedral particles, 40–45 nm in diameter, that enclose a circular dsDNA genome of 3962–7369 bp [2]. The polyomavirus genes are expressed in a time-dependent manner. The products of the early genes are primarily regulatory proteins (e.g., the large and small tumor antigens [LTA and STA]), and the late genes code for the viral proteins (VPs) VP1, VP2, and VP3, which are responsible for virion formation [2, 4,5,6]. Additional coding capacity has been noted; for example, the genome of avian polyomaviruses may encode an X protein or a VP4 protein downstream of the replication origin [2, 5,6,7,8].
Our knowledge about the genetic diversity of mammalian polyomaviruses has increased rapidly over the past 10 years, which has led to an extended taxonomic classification, with more than 100 species. Gammapolyomaviruses are represented by nine species [1,2,3, 7]. Goose hemorrhagic polyomavirus and budgerigar fledgling disease virus, two well-characterized, high-mortality avian polyomaviruses, have been described in a number of bird species that might play a role in their natural circulation [8, 9]. Hence, it seems plausible that wild birds serve as hosts for novel gammapolyomaviruses as well as for some highly pathogenic gammapolyomaviruses associated with economic losses.
In this study, wild birds that died in 2019 at the Zoo and Botanical Garden, Budapest (Hungary), were tested for polyomaviruses. Approximately 50-100 mg of internal organ tissue samples were homogenized in PBS using a TissueLyzer LT instrument (QIAGEN, Hilden, Germany) and were centrifuged for 10,000 × g for 5 min. Nucleic acid was extracted using a ZiXpress-32® Automated Nucleic Acid Purification Instrument and a ZiXpress-32® Viral Nucleic Acid Extraction Kit (Zinexts Life Science Corp., New Taipei City, Taiwan) from a mixture of the prepared samples from each bird.
Polyomavirus DNA was detected using a broad-spectrum nested PCR assay with the primer sets VP1-1f and VP1-1r, and VP1-2f and VP1-2r, described by Johne and co-workers [10]. Sequencing of the amplicons revealed traces of polyomavirus sequence in one out of 32 specimens collected from kidney and liver samples from a great cormorant (Phalacrocorax carbo). The bird was admitted to the zoo’s rescue station with presumed traumatic injuries, but detailed pathological findings were not available.
The back-to-back PCR primers PyV_20190702-2_F (5’-TGGGAAGATGTACTATAGGGGTCTTC-3’) and PyV_20190702-2_R (5’-TCTGACTGCACAACAAACCCAC-3’) (annealing at 65 °C) were designed for amplification of the circular polyomavirus genome using Phusion DNA polymerase according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). The PCR product was purified and prepared for next-generation sequencing using an Illumina NextSeq™ 500 platform as described elsewhere [11]. Sequence contigs were obtained by de novo assembly using Geneious Prime® v.2020.2.4 (Biomatters, Auckland, New Zealand) and CLC Genomics Workbench v9 (QIAGEN, Hilden, Germany) software. The ORFs were predicted using the Open Reading Frame Finder tool (https://www.ncbi.nlm.nih.gov/orffinder/). The sequences were edited using AliView software and were aligned using the MAFFT algorithm implemented in the Geneious Prime software [12]. Maximum-likelihood phylogenetic analysis was performed using PhyML software (LG+G+I+F model, aLRT SH-like branch support) using reference sequences [1, 13]. The phylogenetic tree was visualized and edited using MEGA6 software [14].
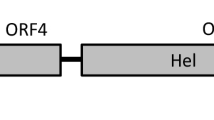
Altogether, 528,035 sequence reads mapped to the novel genome with a sequencing depth of > 3100. The priming site of the back-to-back PCR was determined by Sanger sequencing, resulting in lower sequencing depth in this region. The novel genome (GenBank accession number MZ666388) was found to be 5133 bp long, and the genomic structure resembles that of gammapolyomaviruses, containing the putative ORFs encoding the LTA, STA, VP1, VP2, and VP3 proteins (Fig. 1, Table 1) [1,2,3,4,5,6,7,8]. Furthermore, ORF-X was predicted upstream of the VP2. We observed signatures of mRNA splicing in both the LTA and ORF-X genes (Fig. 1, Table 1). Analysis of representative complete genome sequences of members of all polyomavirus species, performed using RDP4 software, did not reveal any recombination events affecting the genome of the cormorant polyomavirus (CoPyV) [15].

(A) Schematic representation of the genomic structure of cormorant polyomavirus. (B) Maximum-likelihood phylogenetic tree of LTA aa sequences of polyomaviruses constructed using PhyML software, applying the GTR+G+I model and aLRT SH-like branch support. Branches with < 80 support were hidden. The sequence of Japanese eel endothelial cells-infecting virus (GenBank accession number AB543063) was used to root the tree. The cormorant polyomavirus (Phalacrocorax carbo polyomavirus 1) is indicated by a blue triangle
Typical motifs similar to those in other avian polyomaviruses could be identified in the LTA of CoPyV, including the polyomavirus conserved region (CR1, LEELL), the hexapeptide in the J domain (HPDKGG), the pRB1-binding motif (LHAEE), the nuclear localization signal (NLS, TPPKDRAT), the zinc finger motif (CETCKAQKKDMPFRMLKRKWVGGHIDDH), and the ATPase motifs (GGVNTGKT and GAVPVNLE) [4, 6, 7]. The VP3 started with an in-frame methionine of the VP2, as part of the motif MALMPY, which conformed to the consensus motif MALXXΦ (Φ = W, F, Y) described also for other polyomaviruses [4]. The C-terminal region of the putative VP2 and VP3 proteins of CoPyV and all other gammapolyomaviruses is rich in arginine (R) and lysine (K), which may be components of functional NLSs [4]. Although NLSs have been recognized in VP1 proteins of mammalian polyomaviruses [4], an accumulation of basic amino acids is not typical for this region of avian polyomaviruses.
In the LTA-based phylogenetic tree, the CoPyV sequence branched together with gammapolyomavirus sequences (Fig. 1). Each of the main coding sequence of CoPyV and the gammapolyomaviruses shared a maximum of 62.5% nt and 66.6% aa sequence identity in pairwise comparisons, showing the highest values with sequences from goose hemorrhagic polyomavirus, Adélie penguin polyomavirus, and butcherbird polyomavirus. According to the demarcation criteria for polyomaviruses, including a genetic distance of > 15% for the LTA aa sequence [3], CoPyV may be the first member of a novel species within the genus Gammapolyomavirus, for which we propose the name “Phalacrocorax carbo polyomavirus 1”.
Data availability
The sequence data are available in the GenBank database with accession number MZ666388.
References
Ehlers B, Anoh AE, Ben Salem N, Broll S, Couacy-Hymann E, Fischer D, Gedvilaite A, Ingenhütt N, Liebmann S, Martin M, Mossoun A, Mugisha L, Muyembe-Tamfum JJ, Pauly M, Pérez de Val B, Preugschas H, Richter D, Schubert G, Szentiks CA, Teichmann T, Walter C, Ulrich RG, Wiersma L, Leendertz FH, Calvignac-Spencer S (2019) Novel polyomaviruses in mammals from multiple orders and reassessment of polyomavirus evolution and taxonomy. Viruses 11(10):930. https://doi.org/10.3390/v11100930
Moens U, Krumbholz A, Ehlers B, Zell R, Johne R, Calvignac-Spencer S, Lauber C (2017) Biology, evolution, and medical importance of polyomaviruses: an update. Infect Genet Evol 54:18–38. https://doi.org/10.1016/j.meegid.2017.06.011
Polyomaviridae Study Group of the International Committee on Taxonomy of Viruses, Calvignac-Spencer S, Feltkamp MCW, Daugherty MD, Moens U, Ramqvist T, Johne R, Ehlers B (2016) A taxonomy update for the family Polyomaviridae. Arch Virol 161(6):1739–1750. https://doi.org/10.1007/s00705-016-2794-y
Ehlers B, Moens U (2014) Genome analysis of non-human primate polyomaviruses. Infect Genet Evol 26:283–294. https://doi.org/10.1016/j.meegid.2014.05.030
Kaszab E, Szabadi L, Kepner A, Bajnóczi P, Lengyel G, Bányai K, Fehér E (2021) Viral gene expression profile of goose haemorrhagic polyomavirus in susceptible primary cell. Avian Pathol (accepted manuscript).
Johne R, Müller H (2007) Polyomaviruses of birds: etiologic agents of inflammatory diseases in a tumor virus family. J Virol 81(21):11554–11559. https://doi.org/10.1128/JVI.01178-07
Varsani A, Porzig EL, Jennings S, Kraberger S, Farkas K, Julian L, Massaro M, Ballard G, Ainley DG (2015) Identification of an avian polyomavirus associated with Adélie penguins (Pygoscelis adeliae). J Gen Virol 96(4):851–857. https://doi.org/10.1099/vir.0.000038
Kaszab E, Marton S, Erdélyi K, Bányai K, Fehér E (2021) Genomic evolution of avian polyomaviruses with a focus on budgerigar fledgling disease virus. Infect Genet Evol 90:104762. https://doi.org/10.1016/j.meegid.2021.104762
Stys-Fijol N, Kozdrun W, Czekaj H (2016) Preliminary survey of the occurrence of goose haemorrhagic polyomavirus (GHPV) in wild birds in Poland. J Vet Res 60:135–139. https://doi.org/10.1515/jvetres-2016-0019
Johne R, Enderlein D, Nieper H, Müller H (2005) Novel polyomavirus detected in the feces of a chimpanzee by nested broad-spectrum PCR. J Virol 79(6):3883–3887. https://doi.org/10.1128/JVI.79.6.3883-3887.2005
Bali K, Bálint Á, Farsang A, Marton S, Nagy B, Kaszab E, Belák S, Palya V, Bányai K (2021) Recombination events shape the genomic evolution of infectious bronchitis virus in Europe. Viruses 13(4):535. https://doi.org/10.3390/v13040535
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. https://doi.org/10.1093/bioinformatics/btu531
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321. https://doi.org/10.1093/sysbio/syq010
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1(1):vev003. https://doi.org/10.1093/ve/vev003
Funding
Open access funding provided by Veterinary Medical Research Institute. This research was funded by the Momentum program (Hungarian Academy of Sciences), grant number LP2011-10.
Author information
Authors and Affiliations
Contributions
K.B. (Krisztián Bányai) and E.F. designed the study. E.S. and M.H. provided samples and data. E.F., E.K., K.B. (Krisztina Bali) performed experiments and data analysis. E.F., E.K. and K.B. (Krisztián Bányai) prepared the first manuscript draft. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
The authors confirm that no ethical approval was required.
Additional information
Handling Editor: Graciela Andrei .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fehér, E., Kaszab, E., Bali, K. et al. A novel gammapolyomavirus in a great cormorant (Phalacrocorax carbo). Arch Virol 167, 1721–1724 (2022). https://doi.org/10.1007/s00705-022-05478-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05478-8