
Abstract
Norovirus is recognized as one of the leading causes of acute gastroenteritis outbreaks. Genotype GII.9 was first detected in Norfolk, VA, USA, in 1997. However, the complete genome sequence of this genotype has not yet been determined. In this study, a complete genome sequence of GII.9[P7] norovirus, SCD1878_GII.9[P7], from a patient was determined using high-throughput sequencing and rapid amplification of cDNA ends (RACE) technology. The complete genome sequence of SCD1878_GII.9[P7] is 7544 nucleotides (nt) in length with a 3’ poly(A) tail and contains three open reading frames. Sequence comparisons indicated that SCD1878_GII.9[P7] shares 92.1%-92.3% nucleotide sequence identity with GII.P7 (AB258331 and AB039777) and 96.7%-97.4% identity with GII.9 (AY038599 and DQ379715). The results suggested that SCD1878_GII.9[P7] is a member of P genotype GII.P7 and G genotype GII.9. This viral sequence fills a gap at the whole-genome level for the GII.9 genotype.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Norovirus (NoV) is recognized as one of the leading causes of acute gastroenteritis outbreaks. NoV belongs to the family Caliciviridae and has a positive-sense ~7.5 kb RNA genome [1]. Phylogenetically, NoV can be segregated into 10 genogroups and further divided into genotypes based on amino acid sequence diversity in the VP1 gene. GII is the largest of the known genogroups, consisting of 26 genotypes, including 23 human NoV genotypes that are responsible for most epidemics, and three porcine NoV genotypes (GII.11/18/19) [2]. As the diversity of NoV increased through recombination, dual typing was proposed for NoV classification. Partial nucleotide sequences of the RNA-dependent RNA polymerase (RdRp) region of ORF1 are used for NoV P-type classification independently from genotype. A total of 37 P-types have now been identified for in GII viruses [2].
The first strain of genotype GII.9 virus (VA97207) was detected in Norfolk, VA, USA, in 1997 [3]. A partial genome sequence of this strain (a 3290-bp fragment including the complete ORF2 region) was uploaded to the GenBank database in 2001 (accession number AY038599) [3]. Compared with other genotypes, GII.9 strains have rarely been reported. Gelaw et. al. detected only one GII.9 strain in 450 clinical samples by RT-PCR and partially sequenced its VP1 gene (300 bp) [4]. The presence of GII.9 was also reported in wastewater in South Africa and oyster samples in Japan [5, 6]. Nevertheless, there was no submission of a GII.9 sequence to NoroNet from 2005 to 2016 [7].
Materials and methods
In this study, a rare GII.9[P7] whole genome sequence was obtained from a clinical sample. An anal swab and epidemiological data were collected through the acute gastroenteritis (AGE) outbreak surveillance system monitored by Shanghai Customs. The patient was a 22-year-old Japanese female who traveled from India and arrived in Shanghai Pudong Airport on March 19, 2018. The patient had diarrhea and vomiting and was diagnosed as having AGE.
The majority of the whole viral sequence was determined using RNA-seq, and the ends of the viral genome were sequenced using a rapid amplification of cDNA ends (RACE) kit (Vazyme, Nanjing, China) (Supplementary Figs. S1 and S2) [8, 9]. The whole genomic sequence was then assembled and validated using CLC Genomics Workbench (https://digitalinsights.qiagen.com). The assembled viral genome sequence was genotyped using a web-based genotyping tool [10], and a phylogenetic tree was constructed using MEGA X [11]. The complete sequence, named SCD1878_GII.9[P7], was deposited in the GenBank database with the accession number MZ312111.
A total of 1976 human NoV genome sequences (6400-8500 bp) were obtained from ViPR on March 10, 2021 [12]. BioAider was used to remove sequences with sequence identity over 97% [13]. PhyloSuite was used to conduct, manage, and streamline the analyses [14]. Sequences were aligned using MAFFT [15]. The best partitioning scheme and evolutionary models for one pre-defined partition were selected using PartitionFinder2 [16], using the greedy algorithm and the AICc criterion. Maximum-likelihood phylogenetic trees were constructed using IQ-TREE [17] with the GTR+I+G4+F model and 20000 ultrafast bootstrap replicates, using the Shimodaira-Hasegawa-like approximate likelihood-ratio test [18].
Results and discussion
The complete genome sequence of SCD1878_GII.9[P7] is 7544 nucleotides (nt) in length, with a 3’ poly(A) tail. As expected, the genome contains three open reading frames (ORFs) (Table 1). ORF1 can be cleaved into six nonstructural proteins: p48, NTPase, p22, VPg, Pro, and RdRp. The remaining two ORFs encode two structural proteins (VP1 and VP2). A comparison of the sequence against the reference sequence (NC_029646.1, GII.12[P12]) is summarized in Table 1.
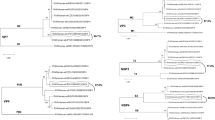
Sequence comparisons indicated that SCD1878_GII.9[P7] shares 92.1%-92.3% and 96.7%-97.4% sequence identity with GII.P7 (AB258331 and AB039777) and GII.9 (AY038599 and DQ379715) at the nucleotide level in the RdRp gene and the amino acid level in the VP1 protein, respectively, suggesting that SCD1878_GII.9[P7] is a member of P genotype GII.P7 and G genotype GII.9 (Fig. 1). To investigate whether this isolate constitutes a new GII.P9 genotype, the RdRp region of DQ379715, AY038599 (GII.9), and reference sequences of GII.[P6]/[P7]/[P20]/[P15] were used to conduct evolutionary analysis by the maximum-likelihood method using the Kimura 2-parameter model. According to the "2-standard-deviation" (SD) criterion, where “the average distance between all sequences within a new genogroup or genotype and its nearest established cluster(s) should not overlap within 2 SD”, an overlap was observed between the average distance of this sequence and P6 or P7 sequences. Thus, the RdRp region of the related GII.9[P7] sequence could not form a new cluster in the phylogenetic tree, and the criterion of 2×SD could not be fulfilled [19, 20]. No significant difference was observed, and therefore, it could not be recognized as a new P type (Supplementary Fig. S3).

Phylogenetic tree of genotypes (left) and P-types (right) based on amino acid sequences of the complete VP1 protein and nucleotide sequences of the RNA-dependent RNA polymerase (RdRp) region respectively. The percentage of replicate trees (>75%) in the bootstrap test (500 replicates) is shown next to the branches.
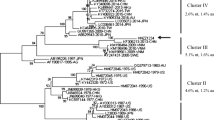
Phylogenetic analysis of whole genome sequences showed that SCD1878_GII.9[P7] clustered into a monophyletic clade with high confidence (bootstrap value = 100%, Fig. 2), together with three genotypes: GII.6[P7], GII.7[P7], and GII.14[P7]. Within the clade, SCD1878_GII.9[P7] formed its own distinct branch, confirming this sequence to be the first whole genome sequence of a GII.9[P7] genotype isolate. Potential recombination within the viral genome was screened using SimPlot, and no evidence for recombination events was detected in the genome of SCD1878_GII.9[P7] (Supplementary Fig. S4) [21].

Maximum-likelihood phylogenetic tree for human NoV genome sequences (6400-8500 bp). The overall evolutionary relationship of SCD1878_GII.9[P7] to closely related NoV genogroups is shown in the tree on the left. An enlarged view of SCD1878_GII.9[P7]-related sequences is shown for the portion of the tree indicated by a yellow box. Ultrafast bootstrap values and Shimodaira-Hasegawa-like approximate-likelihood ratios are included in the node labels.
The rapid development of sequencing technology has greatly facilitated virus monitoring. With the development of second- and third-generation sequencing technologies, discovering and analyzing longer viral genomes has become practical. Additional complete RdRp sequences or, ideally, complete genome sequences for all reference strains will help to improve the robustness of the present classification system [19]. Obtaining whole genome sequences of rare genotypes will not only enrich the database but also provide valuable information for analysis of evolution, as well as reference genome sequences for analysis of diversity, and screening for drug and vaccine development.
Nucleotide sequence accession number The GenBank accession number for norovirus SCD1878_GII.9[P7] is MZ312111.
References
Vinjé J, Estes MK, Esteves P, Green KY, Katayama K, Knowles NJ, Homme YL, Martella V, Vennema H, White PA (2019) ICTV Virus Taxonomy profile: caliciviridae. J Gen Virol 100:1469–1470
Chhabra P, Graaf MD, Parra GI, Chan MC, Green K, Martella V, Wang Q, White PA, Katayama K, Vennema H, Koopmans MPG, Vinjé J (2020) Corrigendum: updated classification of norovirus genogroups and genotypes. J Gen Virol 101:893–893
Jiang X, Zhong WM, Farkas T, Huang PW, Wilton N, Barrett E, Fulton D, Morrow R, Matson DO (2002) Baculovirus expression and antigenic characterization of the capsid proteins of three Norwalk-like viruses. Arch Virol 147:119–130
Gelaw A, Pietsch C, Mann P, Liebert UG (2019) Molecular detection and characterisation of sapoviruses and noroviruses in outpatient children with diarrhoea in Northwest Ethiopia. Epidemiol Infect 147:e218
Mabasa VV, Meno KD, Taylor MB, Mans J (2018) Environmental surveillance for noroviruses in selected South African wastewaters 2015–2016: emergence of the novel GII.17. Food Environ Virol 10:16–28
Imamura S, Kanezashi H, Goshima T, Suto A, Ueki Y, Sugawara N, Ito H, Zou B, Kawasaki C, Okada T, Uema M, Noda M, Akimoto K (2018) Effect of high pressure processing on a wide variety of human noroviruses naturally present in aqua-cultured Japanese oysters. Foodborne Pathog Dis 15:621–626
van Beek J, de Graaf M, Al-Hello H, Allen DJ, Ambert-Balay K, Botteldoorn N, Brytting M, Buesa J, Cabrerizo M, Chan M, Cloak F, Di Bartolo I, Guix S, Hewitt J, Iritani N, Jin M, Johne R, Lederer I, Mans J, Martella V, Maunula L, McAllister G, Niendorf S, Niesters HG, Podkolzin AT, Poljsak-Prijatelj M, Rasmussen LD, Reuter G, Tuite G, Kroneman A, Vennema H, Koopmans M (2018) Molecular surveillance of norovirus, 2005–16: an epidemiological analysis of data collected from the NoroNet network. Lancet Infect Dis 18:545–553
Liu D, Zhang Z, Li S, Wu Q, Tian P, Zhang Z, Wang D (2020) Fingerprinting of human noroviruses co-infections in a possible foodborne outbreak by metagenomics. Int J Food Microbiol 333:108787
Liu D, Zhang Z, Wu Q, Tian P, Geng H, Xu T, Wang D (2020) Redesigned duplex RT-qPCR for the detection of GI and GII human noroviruses. Engineering-PRC 6:442–448
Kroneman A, Vennema H, Deforche K, Avoort HVD, Peñaranda S, Oberste MS, Vinjé J, Koopmans M (2011) An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol 51:121–125
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, Zhou L, Larson CN, Dietrich J, Klem EB, Scheuermann RH (2012) ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 40:D593–D598
Zhou Z, Qiu Y, Pu Y, Huang X, Ge X (2020) BioAider: An efficient tool for viral genome analysis and its application in tracing SARS-CoV-2 transmission. Sustain Cities Soc 63:102466
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT (2020) PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 20:348–355
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30:1188–1195
Guindon S, Dufayard J, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Chhabra P, de Graaf M, Parra GI, Chan MC, Green K, Martella V, Wang Q, White PA, Katayama K, Vennema H, Koopmans MPG, Vinjé J (2019) Updated classification of norovirus genogroups and genotypes. J Gen Virol 100:1393–1406
Kroneman A, Vega E, Vennema H, Vinjé J, White PA, Hansman G, Green K, Martella V, Katayama K, Koopmans M (2013) Proposal for a unified norovirus nomenclature and genotyping. Arch Virol 158:2059–2068
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Funding
This research was funded by the National Science and Technology Major Project (2018ZX10101003-002), General Administration of Customs Project (2021HK131), and State Key Laboratory of Applied Microbiology Southern China (SKLAM 002-2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Ethical approval for this study was obtained from the China CDC Ethical Review Committee (no. M202007) (Beijing, China).
Additional information
Handling Editor: Reimar Johne.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Liu, D., Zhang, Z. et al. Complete genome sequence of GII.9 norovirus. Arch Virol 167, 249–253 (2022). https://doi.org/10.1007/s00705-021-05257-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05257-x