
Abstract
A temperate siphovirus, phiCDKH01, was obtained from a clinical isolate of Clostridioides difficile. The phage genome is a 45,089-bp linear double-stranded DNA molecule with an average G+C content of 28.7%. It shows low similarity to known phage genomes, except for phiCD24-1. Genomic and phylogenetic analysis revealed that phiCDKH01 is a newly discovered phage. Sixty-six putative ORFs were predicted in the genome, 37 of which code for proteins with predicted functions. The phiCDKH01 prophage was localized in the host genome. The results of this study increase our knowledge about the genetic diversity of tailed phages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clostridioides difficile is a pathogen with great epidemiological potential that poses a serious threat to human health [1]. In the CDC's latest report on the risk of drug resistance, C. difficile was classified as the leading cause of nosocomial infections [2]. C. difficile infection (CDI) is closely related to the weakening of the function of the intestinal microbiome as a side effect of antibiotic therapy [3, 4]. CDI is complex and most often manifested with mild, moderate, or severe diarrhea. The development of CDI infection can turn into life-threatening pseudomembranous colitis or toxic megacolon [5,6,7]. Currently, acute C. difficile infection is treated with antibiotics, e.g., metronidazole, vancomycin, or fidaxomicin [8]. The use of antibiotics in the treatment of CDI increases the risk of exacerbation of microflora dysbiosis, causing a reduction or elimination of normal intestinal commensals. Consequently, C. difficile may colonize this niche [9]. Moreover, in the case of this infection, antibiotic therapy promotes the recurrence of the disease and increases the chance of emergence of antibiotic resistance [10].
In the last decade, interest in bacteriophages that infect the pathogen C. difficile has increased due to their possible contribution to virulence and host biology and their potential as alternative therapeutic agents [11]. So far, all of the phages known to infect C. difficile are temperate. In most cases they were isolated from bacterial cells after induction of prophages [12,13,14]. The described C. difficile phages belong to the family Myoviridae or Siphoviridae of the order Caudovirales, i.e., phages with contractile or non-contractile tails, respectively [12, 15]. Myoviruses are the most numerous, and their genomes show significant DNA sequence similarity, with a tendency to group into phylogenetically related clusters. In contrast, a limited number of siphoviruses have been described and sequenced, and these phages have been shown to be more genetically diverse [16].
In the current study, a newly discovered phage named phiCDKH01 was isolated and characterized. The phage genome was sequenced and annotated, and phylogenetic analysis indicated that phiCDKH01 is a member of the family Siphoviridae and might belong to a novel phage lineage. We also determined the location of the phiCDKH01 prophage in the genome of its host.
Bacterial strain isolation
Clostridioides difficile CD34-Sr was isolated from a hospital environment, in the 600-bed clinical hospital of the Medical University of Silesia, Katowice, Poland. The strain was isolated from a bed frame in a patient's room of a nephrology ward. The material was collected using a selective broth enabling the germination of C. difficile spores (C diff Banana Broth, Hardy Diagnostics, Santa Maria, USA). After incubation, one loop of broth was replated on the selective C. difficile medium chromID C. difficile (bioMérieux, Marcy L'Etoile, France) and incubated for 48 hours under anaerobic conditions. Colonies with a characteristic horse odor and yellow-green fluorescence under UV light, microscopically recognized as long, irregular cells with a bulge at their terminal ends, were identified as C. difficile using an automated system (VITEK 2 Compact, bioMérieux, Marcy L'Etoile, France).
Prophage induction and phage isolation
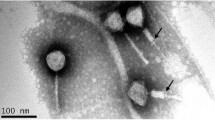
To determine if strain CD34-Sr contained a functional prophage, we used the mitomycin C high-throughput induction method described previously [14]. In this method, the inducible phage DNA in the heated lysate is identified by PCR using specific phage primers targeting the holin genes of myoviruses and siphoviruses [17]. The results confirmed that the amplified PCR product was from the holin gene of an induced siphovirus. We therefore used C. difficile CD34-Sr for large-scale phage induction. Mitomycin C induction was performed on 500 ml of log-phase bacteria cultured in BHI broth (Sigma-Aldrich, St. Louis, USA). Following the overnight incubation, the phage lysate was collected, filtered, and concentrated using polyethylene glycol precipitation [18]. We analyzed the concentrated phage lysate under an electron microscope and found only one type of phage particle (Supplementary Fig. S1). The phage fraction was then purified by CsCl gradient centrifugation as described previously [18]. The isolated phage was named phiCDKH01, after its discoverer's initials.
Genome sequencing and annotation
Genomic DNA of phage phiCDKH01 was purified using a Phage DNA Isolation Kit (Norgen Biotek Corp., Thorold, Canada) following the manufacturer’s instructions. Whole-genome sequencing was performed by Genomed S.A. (Warsaw, Poland) on an Illumina MiSeq platform with 764-fold coverage. High-quality paired-end reads were assembled de novo using SPAdes v. 3.13.0 (https://github.com/ablab/spades). The resulting consensus sequence was annotated using myRAST v. 36 (https://rast.nmpdr.org/) [19] and deposited in the GenBank database under accession number MN718463.
The genomic features of phiCDKH01
The genome of phage phiCDKH01 is 45,089 bp in length with a G+C content of 28.7%, which is similar to that of its host C. difficile. In the initial annotation, a total of 66 ORFs were identified as probable protein-coding genes. Fifty-three were located on the positive strand, while only 13 were located on the negative strand. No rRNA or tRNA genes were identified. Thirty-seven genes were assigned a predicted function. The complete phage genome could be divided into functional clusters that encode proteins involved in DNA packaging, head and tail morphogenesis, host cell lysis, and replication (Fig. 1). We identified genes for the terminase large subunit (phiCDKH01_44), terminase small subunit (phiCDKH01_43), tail tape measure protein (phiCDKH01_43), two tail family proteins (phiCDKH01_62/63), pre-neck appendage-like protein (phiCDKH01_65), portal protein (phiCDKH01_45), scaffolding protein (phiCDKH01_51), and capsid protein (phiCDKH01_52).

Features of the phage phiCDKH01 genome. The predicted ORFs and their orientations are represented by arrows. The putative functional assignments are indicated below the ORFs. The functional modules were assigned based on gene annotation and genomic organization and are shown in different colours. The position of the CRISPR region with its repeats and spacers is indicated by diagonal hatching.
Additionally, we detected genes encoding proteins whose presence confirms the temperate nature of phiCDKH01, including a recombinase (phiCDKH01_31), integrase (phiCDKH01_12), antirepressors (phiCDKH01_20/24), and five putative transcriptional regulators (phiCDKH01_07/17/19/22/27), suggesting that the prophage could affect some bacterial functions.
We identified the gene cluster for host cell lysis containing an N-acetylmuramoyl-L-alanine amidase (phiCDKH01_06), a putative holin protein (phiCDKH01_05), and an ImmA/IrrE family metallo-endopeptidase (phiCDKH01_13).
We also found genes involved in DNA replication encoding a DnaD domain protein (phiCDKH01_32), a single-stranded DNA-binding protein (phiCDKH01_33), and two putative PemI proteins (phiCDKH01_10/42). These proteins have been shown to be essential for the autonomous replication of natural plasmids with a low copy number, e.g., R100 [20].
Finally, we identified several additional interesting genes that encode proteins with different functions e.g., an ADP-ribosyltransferase exoenzyme family protein (phiCDKH01_48) that might covalently modify cell actin to alter the physiology of eukaryotic cells in a manner similar to Clostridium botulinum C2 or Clostridium perfringens E iota toxins [21]. A gene coding for a putative lipoprotein (phiCDKH01_60) might play a role in cortex modification, and thus spore germination [22]. Another gene is predicted to encode HicB antitoxin (phiCDKH01_23), a member of a type II toxin-antitoxin system family found in bacteria and archaea and has been shown to be involved in the stress response, virulence, and persistence [23] (Supplementary Table S1).
Other interesting features of the phiCDKH01 genome include a putative CRISPR (clustered regularly interspaced short palindromic repeats) element and a nearby CRISPR array comprising five spacers of 35, 36 or 37 bp (Fig 1, Supplementary Table S2). Analysis of the CRISPR array revealed that none of the spacers target known C. difficile phages. Although spacers 2 (100% identity) and 5 (97.14% identity) have been detected in several other C. difficile genomes, spacers 1, 3 and 4 did not match known sequences (Supplementary Table S2). Of note, no other phages were detected in the strain carrying phiCDKH01, suggesting that the CRISPR array in phiCDKH01 might be active and prevent further infection by phages.
Phylogenetic analysis
The entire genome sequence of phiCDKH01 was included in a multiple alignment together with genomic sequences of 10 other Clostridioides difficile siphoviruses available in the GenBank database. The alignment was performed using Mauve v. 2.3.1 (http://darlinglab.org/mauve/mauve.html) [24], by the progressive Mauve method. The results were visualised using FigTree v. 1.4.4 software (https://github.com/rambaut/figtree) (Fig. 2a). The most closely related phage turned out to be phiCD24-1, which was originally isolated from a clinical isolate exhibiting the 078 PCR ribotype [13, 25]. The sequences of phiCDKH01 and phiCD24-1 share 89% identity and can be considered members of the same genus according to ICTV rules (Fig. 2b).

a Comparative phylogenetic analysis base on complete genome sequences of C. difficile siphoviruses available in the GenBank database. The figure represents the guide tree calculated using the progressive Mauve algorithm. Numbers associated with each branch represent node ages. b Comparison of the genome sequence of phage phiCDKH01 (top) with phiCD24-1 (bottom). Predicted ORFs and the direction of transcription are indicated by block arrows. The blue box represents a putative CRISPR element. Conserved regions are shaded in grey. The colour intensity corresponds to sequence identity level (89% to 100%). Genomic comparisons were performed using BLASTn. Similarities with E values lower than 1e-100 are plotted. The figure was produced using Easyfig 2.2.5 [26].
Location of the phiCDKH01 prophage in the genome of C. difficile
Bacterial genomic DNA of strain CD34-Sr was isolated using an E.Z.N.A. Bacterial DNA Kit (Omega Bio-tek, USA). Whole-genome sequencing was performed using an Illumina MiSeq platform (Genomed S.A.) with 72-fold coverage. After a quality check, the reads were assembled de novo in SPAdes v. 3.13.0 to create 70 contigs. These sequences are available in GenBank under accession number JACSDL000000000 and were subjected to automatic annotation. The sequence of phiCDKH01 was found in the contig JACSDL010000003.1 from nt 288,650 to 333,698. The prophage is integrated between the loci H7706_07450 and H7706_07755. H7706_07450 shares sequence similarity with a manganese catalase family protein (GenBank accession no. MBC6710325.1). H7706_07755 is annotated as the ilvB gene, coding for the biosynthetic-type acetolactate synthase large subunit (GenBank accession no.MBC6710385.1).
References
Evans CT, Safdar N (2015) Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis 60(Suppl 2):S66–S71. https://doi.org/10.1093/cid/civ140
Centers for Disease Control and Prevention (2019) Antibiotic resistance threats in the United States. https://doi.org/10.15620/cdc:82532
Kwok CS, Arthur AK, Anibueze CI, Singh S, Cavallazzi R, Loke YK (2012) Risk of Clostridium difficile infection with acid suppressing drugs and antibiotics: meta-analysis. Am J Gastroenterol 107:1011–1019. https://doi.org/10.1038/ajg.2012.108
Slimings C, Riley TV (2014) Antibiotics and hospital-acquired Clostridium difficile infection: update of systematic review and meta-analysis. J Antimicrob Chemother 69:881–891. https://doi.org/10.1093/jac/dkt477
George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, Keighley MR, Alexander-Williams J, Burdon DW (1978) Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br Med J. 1:695. https://doi.org/10.1136/bmj.1.6114.695
Dobson G, Hickey C, Trinder J (2003) Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 29:1030. https://doi.org/10.1007/s00134-003-1754-7
Leffler DA, LaMont JT (2015) Clostridium difficile infection. N Engl J Med. 372:1539–1548. https://doi.org/10.1056/NEJMra1403772
Aslam S, Hamill RJ, Musher DM (2005) Treatment of Clostridium difficile- associated disease: old therapies and new strategies. Lancet Infect Dis 5:549–557. https://doi.org/10.1016/S1473-3099(05)70215-2
Zucca M, Scutera S, Savoia D (2013) Novel avenues for Clostridium difficile infection drug discovery. Expert Opin Drug Discov 8:459–477. https://doi.org/10.1517/17460441.2013.770466
Freeman J, Baines SD, Jabes D, Wilcox MH (2005) Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J Antimicrob Chemother 56:717–725. https://doi.org/10.1093/jac/dki321
Sekulovic O, Fortier L-C (2015) Global transcriptional response of Clostridium difficile carrying the CD38 prophage. Appl Environ Microbiol. 81:1364–1374. https://doi.org/10.1128/AEM.03656-14
Hargreaves KR, Clokie MRJ (2015) A taxonomic review of Clostridium difficile phages and proposal of a novel genus, “Phimmp04likevirus.” Viruses 7:2534–2541. https://doi.org/10.3390/v7052534
Sekulovic O, Garnea JR, Néron A, Fortier L-C (2014) Characterization of temperate phages infecting Clostridium difficile isolates of human and animal origins. Appl Environ Microbiol. 80:2555–2563. https://doi.org/10.1128/AEM.00237-14
Phothichaisri W, Ounjai P, Phetruen T, Janvilisri T, Khunrae P, Singhakaew S, Wangroongsarb P, Chankhamhaengdecha S (2018) Characterization of bacteriophages infecting clinical isolates of Clostridium difficile. Front Microbiol. 9:1701. https://doi.org/10.3389/fmicb.2018.01701
Ackermann HW (2009) Phage classification and characterization. Methods Mol Biol 501:127–140. https://doi.org/10.1007/978-1-60327-164-6_13
Rashid SJ, Barylski J, Hargreaves KR, Millard AA, Vinner GK, Clokie MRJ (2016) Two novel myoviruses from the north of Iraq reveal insights into Clostridium difficile phage diversity and biology. Viruses 8:310. https://doi.org/10.3390/v8110310
Shan J, Patel KV, Hickenbotham PT, Nale JY, Hargreaves KR, Clokie MRJ (2012) Prophage carriage and diversity within clinically relevant strains of Clostridium difficile. Appl Environ Microbiol 78:6027–6034. https://doi.org/10.1128/AEM.01311-12
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T et al (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. https://doi.org/10.1186/1471-2164-9-75
Tsuchimoto S, Ohtsubo H, Ohtsubo E (1988) Two genes, pemK and pemI, responsible for stable maintenance of resistance plasmid R100. J Bacteriol 170:1461–1466. https://doi.org/10.1128/jb.170.4.1461-1466.1988
Popoff MR, Rubin EJ, Gill DM, Boquet P (1988) Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun 56:2299–2306. https://doi.org/10.1128/IAI.56.9.2299-2306.1988
Diaz OR, Sayer CV, Popham DL, Shen A (2018) Clostridium difficile lipoprotein GerS is required for cortex modification and thus spore germination. mSphere. 3:e00205-e218. https://doi.org/10.1128/mSphere.00205-18
Li G, Shen M, Lu S, Le S, Tan Y, Wang J, Zhao X, Shen W, Guo K, Yang Y et al (2016) Identification and characterization of the HicAB toxin-antitoxin system in the opportunistic pathogen Pseudomonas aeruginosa. Toxins. 8:113. https://doi.org/10.3390/toxins8040113
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14:1394–1403. https://doi.org/10.1101/gr.2289704
Fortier L-C, Moineau S (2007) Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl Environ Microbiol 73:7358–7366. https://doi.org/10.1128/AEM.00582-07
Sullivan MJ, Petty NK, Beatson SA (2011) Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. https://doi.org/10.1093/bioinformatics/btr039
Funding
This research was supported by the National Science Centre of Poland MINIATURA Programme (2018/02/X/NZ6/01360).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies performed with human participants or animals.
Additional information
Handling Editor: T. K. Frey.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hinc, K., Kabała, M., Iwanicki, A. et al. Complete genome sequence of the newly discovered temperate Clostridioides difficile bacteriophage phiCDKH01 of the family Siphoviridae. Arch Virol 166, 2305–2310 (2021). https://doi.org/10.1007/s00705-021-05092-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05092-0