
Abstract
Here, we report the development of an indirect enzyme-linked immunosorbent assay (ELISA) method that involves using multiepitope recombinant S protein (rSP) as the coating antigen to detect antibodies against canine coronavirus (CCoV). rSP was designed by arranging its four S fragments (91–135 aa, S1 gene; 377–434 aa, S2 gene; 647–671 aa, S3 gene; 951–971 aa, S4 gene; 207–227 aa) and two T-cell epitopes in tandem: T–E1–E2–E3–E4–T. This multiepitope antigen, which has a molecular weight of approximately 25 kDa and contains a His-tag, was recognized by a CCoV-positive serum in a Western blot assay. The optimal concentration of rSP as a coating antigen in the ELISA was 2 μg/mL, and the optimal dilution of enzyme-labeled secondary antibody was 1:10,000. The cutoff OD450 value was established at 0.2395. No reactivity was observed with antisera against canine distemper virus, canine parvovirus, or feline calicivirus, indicating that this assay is highly specific. We also tested 64 clinical serum samples using our newly established method, and the positive rate was found to be 82.8%. In conclusion, our assay was found to be highly sensitive and specific for the detection of antibodies against CCoV, and it can therefore serve as a new, efficient diagnostic method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Canine coronavirus (CCoV), an enteric virus that mainly causes diarrhea with varying severity, has been described since 1971, and it is associated with high morbidity and low mortality [1,2,3]. In cases of coinfection with other pathogens such as canine adenovirus or canine parvovirus (CPV), CCoV infection can lead to severe clinical signs and can be fatal [4]. In recent years, the number of cases of highly pathogenic CCoV infection has increased considerably [5]; moreover, the antibody-positive rate has also increased each year.
CCoV is an enveloped virus with a single-strand positive-sense RNA genome of approximately 26–32 kilobases [6]. Like other coronaviruses, CCoV contains four main structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N) [7]. The S protein (SP) is a major antigenic determinant, and it induces the production of neutralizing antibodies in the host, mediates host-cell receptor binding and viral entry, and triggers fusion between the viral and host-cell membranes. In addition, it is an important determinant of cell tropism and pathogenicity [8, 9]. CCoV detection primarily relies on serological, etiological, and molecular approaches. Three assays – serum neutralization, indirect immunofluorescence, and enzyme-linked immunosorbent assay (ELISA) – are widely used to detect CCoV-specific antibodies. Serum neutralization is the classical serological detection method, but it is cumbersome and time-consuming. Furthermore, this method needs to be performed in a laboratory with a high biosafety level, and for these reasons, serum neutralization is rarely used for rapid clinical detection [10,11,12]. As reported previously [13], the sensitivity of ELISA for detection of CCoV-specific antibodies is significantly higher than that of the serum neutralization test and can be used as an alternative. Indirect immunofluorescence is another method for detecting CCoV-specific antibodies, but the equipment required is expensive [14]. Thus, at present, ELISA is widely used for serological profiling of CCoV, with most established methods using either the M or N protein as the antigen, as these proteins are highly conserved and immunogenic [15,16,17]. SP reportedly plays a key role in inducing neutralizing antibodies and conferring protection against CCoV [18,19,20,21]. Hence, the use of SP as the antigen in ELISA appears to be especially suitable for CCoV detection. SP is a large protein with considerable hydrophobicity, and it is highly glycosylated, which makes the heterologous expression of full-length SP challenging. We therefore constructed a multiepitope antigen of SP to establish an ELISA-based method to detect antibodies against CCoV. We also tested the sensitivity and specificity of our method.
Materials and methods
Predicting and screening linear B-cell epitopes
Based on the sequence of the gene encoding the SP of CCoV (Table 1), as described previously by Miller et al. and Qiao et al. [22, 23], we predicted and screened linear B-cell epitopes using DNAStar software (protean) and the IEDB website (http://www.iedb.org/). We screened areas where the antigenic index, surface probability, and hydrophilicity values are positive, and where the beta turn area overlaps these areas best. A three-dimensional structural model of the protein was built using SWISS-MODEL (PyMOL).
Expression, purification, and identification of recombinant SP (rSP)
To obtain the recombinant plasmid pET28α-S1, codon optimization was first performed, and the sequences encoding the selected portions of rS1 were synthesized and cloned into the BamHI–XhoI restriction sites of pET28α (+) by BGI (Beijing, China). The recombinant plasmid was then introduced into competent Escherichia coli BL21 (DE3) cells by transformation. The fusion protein was expressed in E. coli BL21 (DE3) cells, positive colonies were selected, and protein expression was induced using 1 mM IPTG. The expression temperatures were optimized (37°C and 16°C), and samples were collected at 8, 12, 16, and 20 h after IPTG induction for analysis by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, 12%). Protein purification was carried out using a His-Tagged Protein Purification Kit (Kangwei Century, Beijing, China) according to the manufacturer's instructions.
Bacterial suspensions were lysed by ultrasonic disruption. The sample was centrifuged, and the pellet was resuspended in binding buffer. The supernatant was collected and passed through a nickel affinity chromatography column. The bound protein was eluted with elution buffer and collected in separate tubes with 0.5 ml per tube. Finally, the target protein was renatured in a gradient of 6 M, 4 M, and 2 M urea buffer solution at 4°C. The purified protein was collected and identified by Western blot and liquid chromatography–mass spectrometry.
The concentration of the recombinant protein was determined using a BCA Protein Assay Kit (Beyotime, Shanghai, China). For mass spectrometry, 10 μg of protein was used. After using proteomics discovery software for mass spectrometry data extraction, we used MASCOT to search a local database using the amino acid sequence of the pET28α-S1 protein.
Establishment of indirect ELISA for detection of CCoV antibodies
Optimization of rSP and serum concentrations
Briefly, rSP diluted in an optimized coating buffer was coated onto 96-well plates. The optimal concentration of rSP was determined by testing various concentrations (0.5, 1, 2, and 4 μg/mL). After coating, the plates were washed three times with PBST (0.05% Tween 20 in PBS) and blocked using an optimized blocking buffer. CCoV-positive and negative sera (PBST was used to obtain 1:50, 1:100, 1:200, and 1:400 dilutions) was then added to rSP-coated wells. A matrix titration was used to determine the optimal concentration of rSP for coating onto the wells and the optimal serum dilution.
Selecting an optimal blocking buffer
We tested 1% BSA, 5% BSA, 5% non-fat milk, and 5% FBS to select an optimal blocking buffer based on the OD450 and P/N values.
Selecting the best secondary antibody incubation conditions
We diluted HRP-labeled mountain rabbit anti-canine IgG (Beijing Solarbio Biotechnology Co., Ltd.) in PBST (1:5000, 1:10,000, 1:15,000, and 1:20,000), and this was then added to each well, followed by incubation for 30, 45, 60, and 75 min. Subsequently, 3,3′,5,5′-tetramethylbenzidine (Beijing Solarbio Biotechnology Co., Ltd.) was added to each well, and the plate was incubated. Absorbance of each well was measured at OD450 using a spectrophotometric ELISA reader, and average values were calculated.
Determination of the cutoff value
Using our optimized conditions for rSP ELISA, 10 serum samples that were identified to be negative using a CCoV Antibody Fluorescence Detection Test Strip (Beijing Kwinbon Biotechnology Co.,Ltd, Beijing, China) were tested. The average S/P and standard deviation were calculated. \(\overline{x }\)± 3S was used as the cutoff value of the rSP ELISA. S/P = (OD450 of samples to be tested - average OD450 of negative samples) / (average OD450 of positive samples - average OD450 of negative samples)
Analysis of the specificity of the rSP ELISA
The specificity of the rSP ELISA was determined using sera from animals that were positive for CCoV and from animals that were positive for canine distemper virus (CDV), CPV, and feline calicivirus (FCV), but negative for CCoV.
Analysis of the sensitivity of the rSP ELISA
For sensitivity analysis, five CCoV-positive serum samples with different antibody levels and a standard CCoV-positive serum sample were diluted (1:100–1:6400) and then subjected to rSP ELISA.
Comparison of rSP ELISA using clinical serum samples
Sixty-four clinical serum samples collected from the Beijing, Hebei, and Shandong regions were subjected to rSP ELISA. The results were compared to those obtained using a Canine Coronavirus Antibody Fluorescence Detection Test Strip (Beijing Kwinbon Biotechnology Co., Ltd, Beijing, China). In this assay, test strips are first coated with the N protein of CCoV, and then with sheep anti-canine IgG fluorescent microspheres. The serum for detection is diluted at 1:2000, and 80 μl of diluted serum is added to the sample well. After 5-10 min, test strips are inserted into a fluorescence immunoassay analyzer (Beijing Kwinbon Biotechnology Co.,Ltd, Beijing, China) for detection, and a fluorescence value greater than or equal to 0.5 is considered positive.
Results
Prediction and screening of B-cell epitopes
Based on the sequence of the gene encoding SP of CCoV, predicted linear B-cell epitopes of SP were chosen. The S domain in the CCoV strain is connected by a KK spacer and the sequence GGGGAS, and rSP consists of four fragments, namely E1 (91–135 aa, S1 gene), E2 (377–434 aa, S2 gene), E3 (647–671 aa, S3 gene), and E4 (951–971 aa, S4 gene), and two T-cell epitopes (Table 1). We screened recombinant fusion proteins and compared their predicted properties, such as antigenicity, hydrophilicity, and surface accessibility. The amino acid sequences of the selected B cell and T cell epitopes were 94.83% to 100% identical to those in the NCBI database. Sequence assembly involved arranging the four fragments in tandem in the following order: T–E1–E2–E3–E4–T. A model of the three-dimensional structure of this protein was made using SWISS-MODEL (PyMOL). The optimized gene was cloned into pET-28α.
Identification of the recombinant plasmid
The constructed recombinant plasmid was subjected to double digestion with BamHI and XhoI, which led to the generation of two bright bands of approximately 5000 bp and 600 bp (Fig. 1). These sizes were consistent with those of the plasmid (5334 bp) and target band (639 bp), respectively.

Agarose gel electrophoresis of the recombinant plasmid pET28α-S1. Lane M, DNA molecular weight marker; lane 1, BamHI and XhoI double-digested product; lane 2, uncut pET28α-S1 plasmid DNA
Expression and identification of the recombinant protein
SDS-PAGE, Western blotting, and purification of rSP
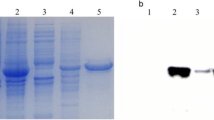
In positive colonies, protein expression was induced using 1 mM IPTG, followed by incubation at 37°C for 8 h. The samples were then subjected to SDS-PAGE. We verified that pET28α-S1 was mainly expressed in an insoluble form in inclusion bodies and was present in large quantities. The size of rSP was approximately 25 kDa (Fig. 2).

Protein expression, purification, and identification. (A) Induction of expression of the recombinant protein. M, protein molecular weight marker; lane 1, protein expression induced by 1 mM IPTG; lane 2, protein expression without IPTG induction; lane 3, expression of the target protein in the supernatant; lane 4, expression of the target protein in inclusion bodies. (B) Analysis of the purified protein using affinity chromatography. M, protein molecular weight marker; lane 1, impurities in the nickel affinity column eluted by binding buffer; lanes 2-5, target protein after purification by nickel affinity chromatography. (C) The protein after renaturation. M, protein molecular weight marker; lane 6, purified rSP. (D) Identification of the recombinant protein via Western blotting. M, protein Molecular Weight Marker. Lane 1, target protein identified by Western blotting
Liquid chromatography – mass spectrometry
Using mass spectrometry analysis, the pertinent peptide was identified to be the purified target protein. The peptides identified were NDVQQRPLLK, YLGTLPPSVKEIAISK, and SLYVIYEEGDNIVGGGGGASILPSHNSK (Fig. 3).

Mass spectrometry results. A. Results pertaining to the peptide NDVQQRPLLK. B. YLGTLPPSVKEIAISK. C. SLYVIYEEGDNIVGGGGGASILPSHNSK
Establishment of indirect ELISA for detecting CCoV antibodies
Using a matrix titration test, the optimal concentration of the antigen rSP was found to be 2 μg/mL, and the optimal serum dilution was 1:200 (Table 2); 5% non-fat milk was the best blocking buffer, with the best incubation temperature being 37°C, with an incubation time of 60 min. Furthermore, ELISA test results were optimal when the secondary antibody was diluted to 1:10,000, the color development time was 10 min, and the incubation temperature was 37°C.
Determination of the cutoff value
The average OD450 values of negative and positive sera were 0.2117 and 1.6611; the average S/P of negative serum samples (\(\overline{x }\)) was 0.1315, and the standard deviation was 0.0360. The cutoff value was 0.1315 ± 3 × 0.0360 = 0.2395 and 0.1315 ± 2 × 0.0360 = 0.2035, respectively. When the S/P value was ≥0.2395, the sample was considered positive, and when the S/P value was ≤0.2035, the sample was considered negative. If the S/P value was between these two values, the sample was considered negative. If the result was ambiguous, the test was repeated, and if the S/P value was still ≤0.2035, the sample was regarded as negative.
Specificity of the rSP ELISA
We used established methods to test CCoV, CDV, canine adenovirus, CPV, and FCV-positive sera, and data were analyzed statistically. The S/P values of CDV-, CPV-, and FCV-positive sera were all lower than the critical value; no cross-reactivity was observed with S1 antigen, and the specificity was good (Fig. 4).

Specificity analysis of rSP ELISA
Sensitivity of the rSP ELISA
Sensitivity analysis indicated that the titer of the standard CCoV-positive serum was 1:800, indicating that the method had good sensitivity (Fig. 5).

Sensitivity analysis of rSP ELISA
Comparison of rSP ELISA with clinical serum samples
For the 64 clinical serum samples obtained from Beijing, Hebei, and Shandong regions, the positive rate was 84.37%. When our indirect rSP ELISA method was used, the positive rate was 82.81%. The total coincidence rate was 98.44% (Table 3).
Discussion
ELISA is a convenient technique for evaluating the response to vaccination, particularly in the case of viruses such as CCoV that have various serotypes, because this type of assay can identify different antibodies via coated antigens. Usually, the coating antigen is either an inactivated virus or a recombinant protein. In recent years, multiple antigens or synthetic peptides have been used for ELISA, and such methods have shown improved sensitivity and specificity. In this study, one of the reasons for using CCoV SP in the form of a multiepitope antigen was to assess whether it can serve as an alternative to full-length SP. SP is encoded by ORF2, has multiple glycosylation sites, exists in the form of a trimer, is located in the outermost layer of the coronavirus particle, and is the main protein of the viral envelope [24, 25]. Because SP is very large, it is difficult to express full-length SP. Using bioinformatics software, we screened all of the dominant epitopes, using epitopes originally described in other studies as references [26,27,28,29]. In this manner, we synthesized the entire gene encoding the epitope, thereby overcoming the issues associated with the high molecular weight of SP and its potential weak reactogenicity. Different serotypes and genotypes of CCoV several epitopes in common, as well as some unique epitopes, and these are of great significance for cross-immunization.
The selected epitope was constructed using pET28α, and competent E. coli BL21 (DE3) cells were used for expression. At 37°C, expression was observed in the form of inclusion bodies. We attempted to lower the culture temperature to 20°C to obtain soluble protein; however, soluble protein was not obtained, although we found that the expression levels at the two temperatures were similar. Western blotting revealed that rSP showed good antigenicity.
In conclusion, we have successfully established an ELISA-based method using multiepitope rSP instead of full-length SP as the coating antigen. This method showed good sensitivity and specificity for detection of antibodies against CCoV, and it can thus serve as a new, efficient diagnostic assay. We believe that our technique can be used in further studies for developing genetic recombinant vaccines and therapeutic monoclonal antibodies.
References
Kaneshima T et al (2006) The prevalence of a group 2 coronavirus in dogs in Japan. J Vet Med Sci 68(1):21–25
Zicola A et al (2012) Fatal outbreaks in dogs associated with pantropic canine coronavirus in France and Belgium. J Small Anim Pract 53(5)
Binn LN et al (1974) Recovery and characterization of a coronavirus from military dogs with diarrhea. In: Proceedings, annual meeting of the United States Animal Health Association, vol 1974, no 78
Tennant BJ et al (1993) Studies on the epizootiology of canine coronavirus. Vet Rec 132(1)
Wang X et al (2016) Co-circulation of canine Coronavirus I and IIa/b with high prevalence and genetic diversity in Heilongjiang Province, Northeast China. PLoS ONE 11(1):e0146975
Siddell SG, Wege H, Meulen V (1983) The biology of coronaviruses. J Gen Virol:761–776
Wesseling JG et al (1994) Nucleotide sequence and expression of the spike (S) gene of canine coronavirus and comparison with the S proteins of feline and porcine coronaviruses. J Gen Virol 75(Pt 7)
Cavanagh D et al (1994) Revision of the taxonomy of the Coronavirus, Torovirus and Arterivirus genera. Arch Virol 135(1–2):227–237
Gallagher TM, Buchmeier MJ (2001) Coronavirus spike proteins in viral entry and pathogenesis. Virology 279(2):371–374
Osterhaus AD et al (1980) Canine viral enteritis: prevalence of parvo-, corona- and rotavirus infections in dogs in The Netherlands. Tijdschr Diergeneeskd 105(20):181–190
Priestnall SL et al (2007) Serological prevalence of canine respiratory coronavirus in southern Italy and epidemiological relationship with canine enteric coronavirus. J Vet Diagn Invest 19(2):176–180
Priestnall SL et al (2006) Serological prevalence of canine respiratory coronavirus. Vet Microbiol 115(1–3):43–53
Pratelli A et al (2002) Prevalence of canine coronavirus antibodies by an enzyme-linked immunosorbent assay in dogs in the south of Italy. J Virol Methods 102(1–2):67–71
Tuchiya K et al (1987) Plaque assay for canine coronavirus in CRFK cells. Nihon Juigaku Zasshi 49(3):571–573
Zhou YH, Yin JB, Liu L, Yin XP, Li YM, Zhang ZD (2020) Prokaryotic expression of canine coronavirus N protein and preparation of its polyclonal antibodies. Chinese Animal Husbandry & Veterinary Medicine
Elia G et al (2003) Recombinant M protein-based ELISA test for detection of antibodies to canine coronavirus. J Virol Methods 109(2):139–142
de Haan CA, Vennema H, Rottier PJ (2000) Assembly of the coronavirus envelope: homotypic interactions between the M proteins. J Virol 74(11):4967–4978
Gebauer F et al (1991) Residues involved in the antigenic sites of transmissible gastroenteritis coronavirus S glycoprotein. Virology 183(1):225–238
De Diego M et al (1992) Epitope specificity of protective lactogenic immunity against swine transmissible gastroenteritis virus. J Virol 66(11):6502–6508
Vennema H et al (1990) Immunogenicity of recombinant feline infectious peritonitis virus spike protein in mice and kittens. Adv Exp Med Biol 276:217–222
Hasony HJ, Macnaughton MR (1981) Antigenicity of mouse hepatitis virus strain 3 subcomponents in C57 strain mice. Arch Virol 69(1):33–41
Miller et al (2003) Canine coronavirus S gene and uses therefore[P].United States Patent. US No: 6,602,504
Qiao J (2005) Construction, Expression and Immunogenicity of Gene Vaccines and Recombinant Adenovirus of CCV, Jilin University
Lai MM, Cavanagh D (1997) The molecular biology of coronaviruses. Adv Virus Res 48:1–100
Deng X et al (2019) Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses Type I and Type III interferon responses. J Virol 93(8)
Karosiene E et al (2012) NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64(3):177–186
Larsen MV et al (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform 8:424
Livingston B et al (2002) A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol 168(11):5499–5506
Deléage G (2017) ALIGNSEC: viewing protein secondary structure predictions within large multiple sequence alignments. Bioinformatics 33(24):3991–3992
Funding
This study was supported by the Agricultural Science and Technology Innovation Program (ASTIP-IAS15) and the National Key Research and Development Program of China (2016YFD0501003).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None.
Additional information
Handling Editor: Sheela Ramamoorthy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hao, YF., Li, SH., Zhang, GZ. et al. Establishment of an indirect ELISA-based method involving the use of a multiepitope recombinant S protein to detect antibodies against canine coronavirus. Arch Virol 166, 1877–1883 (2021). https://doi.org/10.1007/s00705-021-05072-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05072-4