
Abstract
A dengue virus serotype 1 (DENV-1) epidemic occurred from October to December 2018 in Xishuangbanna, Yunnan, Southwest China, neighboring Myanmar, Laos, and Vietnam. In this study, we investigated the molecular characteristics, evolution, and potential source of DENV from Xishuangbanna. The C (capsid), prM (premembrane), and E (envelope) genes of DENV isolated from 87 serum samples obtained from local patients were amplified and sequenced, and the sequences were evaluated by identification of mutations, phylogenetic and homologous recombination analysis, and secondary structure prediction. Phylogenetic analysis showed that all of the epidemic DENV strains from Xishuangbanna could be grouped in a branch with DENV-1 isolates, and were most similar to the Fujian 2005 (China, DQ193572) and Singapore 2016 (MF314188) strains. When compared with DENV-1SS (the standard strain), there were 31 non-synonymous mutations, but no obvious homologous recombination signal was found. Secondary structure prediction showed that some changes had occurred in a helical region in proteins of the MN123849 and MN123854 strains, but there were few changes in the disordered region. This study reveals the molecular characteristics of the structural genes of the Xishuangbanna epidemic strains in 2018 and provides a reference for molecular epidemiology, infection, and pathogenicity research and vaccine development.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dengue virus (DENV) is a ssRNA virus belonging to the family Flaviviridae that is mainly transmitted by Aedes aegypti and Aedes albopictus mosquitoes [1,2,3,4]. The DENV genome encodes three structural proteins (C, prM and E) and seven non-structural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) [5,6,7]. There are four different serotypes of DENV, and infection with any of the four serotypes can result in extremely severe manifestations [8,9,10]. The serotypes are antigenically distinct although they have up to 65% nucleotide sequence identity [11,12,13]. Dengue was classified as the most important mosquito-borne viral disease worldwide by WHO in 2012. DENV infection usually causes a relatively mild disease called dengue fever (DF), but severe disease, including dengue shock syndrome and dengue hemorrhagic fever, can also occur [6, 8]. DF is mainly prevalent in tropical and subtropical areas, especially in urban and suburban areas. The worldwide incidence of dengue infection has been increasing over the past decades, and about half of the world's population is currently at risk of DENV infection. However, no effective treatment or vaccine is available for DF [14,15,16].
Dengue-like illnesses are common in China, and the first record of DENV infection dates back to 1978 in the city of Foshan in Guangdong Province [17,18,19]. Since then, several large outbreaks of DF have occurred in the southeastern part of China, including Zhejiang, Fujian, Taiwan, Hainan, Yunnan, Guangxi, and Guangdong [17]. Sporadic outbreaks or imported cases of dengue were identified from the 1990s to the 2010s in China [19]. However, in 2012, the number of DENV infections began to increase and peaked in 2014 [9, 10]. Located in southwestern China, Yunnan province, neighboring Myanmar, Laos, and Vietnam, was reported to have an outbreak of 56 cases in 2008 [20], which was the first outbreak of the disease in this region since 1949 [20, 21]. Dengue outbreaks or epidemics were reported annually in Yunnan province from 2013 to 2015. In 2013, there were 1319 confirmed cases of DENV-3 in Xishuangbanna for the first time [22, 23]. Since then, there have been two large-scale dengue outbreaks in 2015 and 2017, with 1132 and 1348 confirmed cases, respectively [24, 25].
In 2018, DF cases were reported in some of the Southeast Asian countries neighboring China, including Vietnam, Laos, Cambodia, Myanmar, Thailand, and Malaysia, and DF cases were also reported in the Chinese cities of Yunnan, Hunan, and Guangzhou. Therefore, understanding the potential sources, molecular characteristics, and evolution of DENV is critical for preventing future outbreaks of DF.
In this study, we investigated the potential sources of the causative agent and analyzed the evolution and characteristics of the structural genes of the epidemic DENV strains from 2018.
Materials and methods
Identification and serotyping of DENV
Serum samples were collected from DF patients in Xishuangbanna, Yunnan, China, followed by viral RNA extraction. Dengue NS1 antigen was detected using a DF NS1 test kit (Blue Cross, Beijing, China). Viral RNA was extracted from 150 µL of serum using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The viral RNA was used for amplification the structural protein genes of DENV.
Reverse transcription PCR (RT-PCR) and sequencing of structural protein genes
A PrimeScript One Step RT-PCR Kit (Takara, Dalian, China) was used according to the manufacturer’s instructions to synthesize cDNA. Standard DENV serotyping primers were then used for PCR, followed by sequencing. A NCBI-BLAST search was used for serotyping. In addition, primer pairs were used to amplify the overlapping fragments from nt 96 to 2420 of the DENV-1 genome. All PCR procedures were performed using Golden Star T6 Super PCR Mix (TSINGKE, Kunming, China) using previously described primers [24]. The cycling parameters were as follows: 98 °C for 2 min, followed by 35 cycles of denaturation at 98 °C for 10 s, annealing at 56 °C for 10 s, and extension at 72 °C for 40 s, and then a final extension at 72 °C for 5 min. The PCR product was analyzed by 1% agarose gel electrophoresis, stained with SYBR Safe DNA Gel Stain Dye (Invitrogen, Eugene, USA), and visualized under UV light. Nucleotide sequences were determined by direct sequencing using an ABI 3730XL sequencer (Macrogen, Seoul, Korea). Briefly, the PCR product was excised from the gel and purified, BDT sequencing was performed, and the sequencing results were analyzed. The resulting sequences were compared to sequences in the GenBank database using NCBI BLAST. The sequences were also compared to that of standard strain DENV1-SS (GenBank® ID:EU848545.1).
Phylogenetic analysis
A multiple sequence alignment of the region encoding the structural proteins was performed using Clustal X, using reference sequences from the GenBank database. The total number of reference strains was 51, and their countries of origin and accession numbers are as follows: standard strains (DV-1SS: EU848545, DENV-2SS: M29095, DENV-3SS: M93130, and DENV-4SS: AF326573); China (KF864667, FJ196845, AY835999, FJ176780, DQ193572, EU359008, MF405201, KC762652, KF971869, KX458014, KY672941, and MG679800); Brazil (JX669474, KP188547 and GU131863); Japan (AB074760); Singapore (EU081262, KX380806, MF314188, and GQ357692); Thailand (JQ922547, AY732477, AY732483, and JN638341); Vietnam (JQ045668 and JQ045660); French Polynesia (DQ672559); Malaysia (JN697057 and EF457905); India (JQ922548 and KJ755855); Indonesia (MH823207); Laos (KC172829 and KC172832); Myanmar (AY726550, AY726553, JF459993, and MG679801); Cambodia (AF309641, HM488255 and KU509260); USA (FJ562106 and EU482591); Haiti (KT279761 and KU509264) and Argentina (KC692514 and KC692495). Phylogenetic trees were constructed using MEGA software (version 6.0) by the maximum-likelihood (ML) method with 1000 bootstrap replications.
Molecular characterization
The sequences of the structural protein genes of the Xishaungbanna 2018 DENV-1 epidemic strain were uploaded to the National Center for Biotechnology Research (NCBI) GenBank database (http://www.ncbi.nim.nih.gov/GenBank/index.html) under the accession numbers MN123810-MN123861. The BioEdit program was used to analyze nucleotide substitutions, and the Sequence Manipulation Suite (SMS, http://www.bio-soft.net/sms/index.html) was used to analyze amino acid sequence mutations. SimPlot and RDP version 5.5 (https://web.cbio.uct.ac.za/darren/rdp.html) were used to identify sites where homologous recombination might have occurred in the structural protein genes of epidemic strains in Xishuangbanna. The detailed procedure for RDP analysis was as follows: open an MEG file, left click the “open” button, click the “options” button at the top of the page, then enter the general page to select the corresponding parameters, and left click the “run” button on the main page to start the reorganization analysis. PredictProtein (https://www.predictprotein.org/) was used to predict the secondary structure of the structural proteins of the DEN-1SS and Xishuangbanna 2018 DENV-1 epidemic strains. Potential protein binding sites and the exposed and buried regions in the structural proteins were also predicted. Potential helical structures were also identified.
Results
Geographical location of the dengue fever epidemic of 2018 and the study design
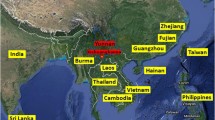
Since the first large-scale outbreak of DF in 2013, there has been an outbreak of this disease with different serotypes every other year in Xishuangbanna. In 2018, there was no large-scale outbreak, only a small-scale epidemic, and DF cases were also reported in some Southeast Asian countries neighboring China (Fig. 1). During the DENV epidemic from October to December 2018 in Xishuangbanna, a total of 87 serum samples from patients were collected at Xishuangbanna Dai Autonomous Prefecture People’s Hospital. All samples were confirmed to be NS1-positive using a colloidal gold test. Viral RNA was successfully extracted from 52 of the samples, followed by gene amplification and sequencing of the region containing the DENV C, prM, and E genes. In order to analyze the origin and genetic relationships of the DENV isolates from Xishuangbanna, phylogenetic analysis was performed and mutations, potential sites of homologous recombination, and secondary structures were predicted.

Geographical relationships of other dengue-endemic countries and regions in 2018. Some of the Southeast Asian countries neighboring China experienced DF in 2018. Red dots represent areas with a dengue epidemic, and a red five-pointed star indicates the location of Xishuangbanna. This figure was drawn using Adobe Illustrator CS6 software.
Phylogenetic analysis
A phylogenetic tree was constructed based on 52 DENV structural protein gene sequences. Reference sequences obtained from the GenBank database, including representatives of the four serotypes of DENV, Chinese epidemic strains, and other epidemic strains from Southeast Asian countries and the rest of the world. The results indicated that all 52 Xishuangbanna epidemic DENV strains were of the DENV-1 serotype and belonged to one branch of the tree. They were most similar to the Fujian 2005 (China, DQ193572) and Singapore 2016 (MF314188) strains. DENV-1 strains have been divided into six genotypes, and the results showed that these epidemic strains belonged to genotype I (Fig. 2). These results indicated that the DENV-1 epidemic in Xishuangbanna in 2018 was caused by a DENV strain circulating in Southeast Asia or a domestic strain.

Phylogenetic analysis of DENV- 1 epidemic strains in Xishuangbanna, Yunnan, China, 2018. The phylogenetic tree was constructed by the maximum-likelihood method with 1000 bootstrap replicates. ○,DENV epidemic strains from Xishuangbanna, Yunnan, China in 2018; The structural protein sequences of the reference strains were obtained from the NCBI database.
Analysis of mutations and recombination sites
All 52 sequences of structural proteins genes were compared to those of the four standard DENV reference strains using BLAST, and it was found that all of them were most similar to DENV-1SS. Since all the sequences were similar, two representative sequences (MN123849 and MN123854) were selected randomly to perform mutation and recombination analysis based on DENV-1SS. A total of 151 nucleotide variations and 31 amino acid sequence differences were observed in the DENV structural protein genes. The mutation rate in the C/prM region was 2.58%, and in the E gene, it was 4.09%. To identify possible sites of homologous recombination in the structural proteins of the two Xishuangbanna 2018 DENV-1 epidemic strains (MN123849 and MN123854), these sequences were compared to that of DENV-1SS using both Simplot and RDP, but no obvious homologous recombination signals were found (Fig. 3).

Comparison of Xishuangbanna 2018 epidemic strains with DENV-1SS. (a) Amino acid substitutions in the MN123849 and MN123854 strains compared with DENV-1SS. The symbol "▲" indicates mutations present in all epidemic strains. (b) Prediction of homologous recombination of the structural protein genes of MN123849 and MN123854 compared with DEN-1SS. No obvious homologous recombination signal was found in these strains.
Secondary structure analysis of structural proteins
Potential secondary structure elements in the structural proteins of the MN123849 and MN123854 strains were predicted and compared with those predicted in DENV-1SS. There were 41 and 42 predicted protein binding sites in MN123849 and MN123854, respectively. Of these, 21 (aa 87, 93, 97, 120, 157, 184, 198, 199, 200, 357, 376, 377, 503, 505, 507, 508, 509, 595, 622, 623 and 642) were found in MN123849, MN123854 and DEN-1SS. However, 9 and 10 sites were lacking in MN123849 (aa 5, 7, 158, 195, 197, 365, 366, 429 and 621) and MN123854 (aa 5, 7, 40, 41, 123, 195, 429, 510, 594 and 596), respectively, compared with DENV-1SS. Two potential nucleotide binding sites (aa 10 and 17) were lacking in MN123849 and MN123854, compared with DEN-1SS. In addition, one nucleotide binding site (aa 98) only found in the MN123849 and MN123854 strains, and two sites (aa 204 and 205) are present only in MN123854. A few changes were observed in disordered regions. Some differences in a predicted helical transmembrane region were observed between DEN-1SS, MN123849, and MN123854 (Fig. 4).

Secondary structure prediction for the structural proteins of Xishuangbanna 2018 epidemic strains. (a) Secondary structure prediction of MN123849 compared with DENV-1SS. (b) Secondary structure prediction of MN123854 compared with DENV-1SS. Yellow dots represent potential DNA-binding regions, black dots represent potential nucleotide-binding regions, purple dots represent potential RNA-binding regions, and red dots represent potential protein-binding regions. Blue and red bands in the first line represent β-strand and α-helical regions, respectively. Blue and yellow bands in the second line represent exposed and buried regions, respectively. Purple bands in the third line indicate helical transmembrane regions, and green bands in the fourth line represent disordered regions.
Discussion
In this study, 87 serum samples were collected from febrile patients in Xishuangbanna. However, only 52 of these samples were successfully amplified by PCR and sequenced. This might have been due to samples being collected at different time points. Phylogenetic analysis was performed based on the structural protein genes (C/prM/E), which are critical for viral adsorption, penetration, replication, and release [26,27,28], and these genes were compared with reference sequences from all over the world. Our results showed that all 52 strains were DENV-1 and belonged to one cluster. They were evolutionarily close to the Fujian (2005) and Singapore (2016) strains. Previous studies have shown that imported cases in China were primarily from Asian countries, and Laos and Myanmar were the main sources of the DF epidemic in Yunnan Province [22, 23, 29, 30]. In addition, our previous study showed that the first DENV-1 outbreak in Xishuangbanna in 2017, including imported cases from Myanmar, was caused by viruses that were closely related to domestic epidemic strains (Guangzhou, JQ048541 and Hubei, KP772252) and Southeast Asia epidemic strain (Laos, KC172834). Compared to the DENV-1 outbreak in 2017, the DENV epidemic in Xishuangbanna in 2018 was small in scale, but the viral strains were again closely related to a domestic epidemic strains (Fujian, DQ193572) and an epidemic strain from Southeast Asia (Singapore, MF314188). This suggests that the outbreak of DF might have been caused by viruses imported from neighboring countries in Southeast Asia or by circulating domestic epidemic strains.
Genetic recombination is a vital mechanism by which viruses evolve and gain genetic variation, and the rate of gene recombination is higher than that of mutation and natural selection alone [31, 32]. To assess whether genetic recombination had occurred in the region encoding the structural proteins of the Xishuangbanna 2018 epidemic strains MN123849 and MN123854, their sequences were compared with that of DENV-1SS, and no evidence of recombination was found.
Although DENV has spread rapidly worldwide and led to millions of people being infected each year, licensed vaccines or effective antiviral treatments are not available to control DENV. Therefore, it is urgent to develop a safe and effective vaccine against DENV infection. Structural proteins, especially the E protein, serve as the main targets in the development of subunit vaccine candidates [33]. The E protein has three domains (I-III). Domain III contains serotype-specific epitopes that can elicit neutralizing antibodies and a host cell receptor recognition site that allows viral attachment and entry into the host cell [34,35,36]. This domain is widely used in vaccine development because it can induce antibodies against DENV infection [36,37,38]. Kawano et al. reported that conversion of the amino acid residue located at position E155 in domain I of the DENV-4 E protein from T to I altered the virulence of the virus [39]. In our previous study, we made a similar observation [24]. In this study, we also observed mutations at amino acid residue 155 (nt position: 435, from T to S) in the E proteins of Xishuangbanna epidemic strains. However, there were no related clinical characteristics of dengue hemorrhagic fever or dengue shock in these patients, almost all of whom had only DF. Therefore, it remains to be determined whether the mutation of amino acid residue 155 (from T to S) affects the virulence of the endemic virus.
Due to geographical location and national economic policy, most Southeast Asian countries have experienced DF epidemic, so it is difficult to find the exact source of the Xishuangbanna 2018 epidemic strains. This study may provide the basis for further study on the molecular epidemiology, infection, pathogenicity, and vaccine development of DENV.
Availability of data and materials
The sequences of the structural protein genes of DENV from Xishaungbanna in this study were uploaded to the GenBank database with the accession numbers MN123810-MN123861.
Abbreviations
- DENV:
-
dengue virus
- DF:
-
Dengue fever
- E:
-
Envelope
- M:
-
Membrane
- C:
-
Capsid
- prM:
-
Premembrane
- DENV-1SS:
-
DENV-1 standard strain
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- ML:
-
Maximum likelihood
- NCBI:
-
National Center for Biotechnology Information
- RDP:
-
Recombination Detection Program
References
Pooja S, Sabeena S, Revti B, Sanjay R, Anjali A, Rajendra K, Aswathyraj S, Giselle D, Hindol M, Arunkumar G (2017) Circulating genotypes of Dengue-1 virus in South West India, 2014–2015. Jpn J Infect Dis 70(6):663–665. https://doi.org/10.7883/yoken.JJID.2017.087
Chambers TJ, Hahn CS, Galler R, Rice CM (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 44:649–688. https://doi.org/10.1146/annurev.mi.44.100190.003245
Ahamed SF, Rosario V, Britto C, Dias M, Nayak K, Chandele A, Kaja MK, Shet A (2019) Emergence of new genotypes and lineages of dengue viruses during the 2012–15 epidemics in southern India. Int J Infect Dis. 84S:S34–S43. https://doi.org/10.1016/j.ijid.2019.01.014
Villabona-Arenas CJ, Zanotto PM (2013) Worldwide spread of Dengue virus type 1. PLoS ONE 8(5):e62649. https://doi.org/10.1371/journal.pone.0062649
Afreen N, Naqvi IH, Broor S, Ahmed A, Kazim SN, Dohare R, Kumar M, Parveen S (2016) Evolutionary analysis of dengue serotype 2 viruses using phylogenetic and Bayesian methods from New Delhi, India. PLoS Negl Trop Dis 10(3):e0004511. https://doi.org/10.1371/journal.pntd.0004511
Avilés G, Meissner J, Mantovani R, St Jeor S (2003) Complete coding sequences of dengue-1 viruses from Paraguay and Argentina. Virus Res. 98(1):75–82. https://doi.org/10.1016/j.virusres.2003.08.018
Ayolabi CI, Olusola BA, Ibemgbo SA, Okonkwo GO (2019) Detection of Dengue viruses among febrile patients in Lagos, Nigeria and phylogenetics of circulating Dengue serotypes in Africa. Infect Genet Evol. 75:103947. https://doi.org/10.1016/j.meegid.2019.103947
Bäck AT, Lundkvist A (2013) Dengue viruses—an overview. Infect Ecol Epidemiol. 30:3. https://doi.org/10.3402/iee.v3i0.19839
Barban V, Munoz-Jordan JL, Santiago GA, Mantel N, Girerd Y, Gulia S, Claude JB, Lang J (2012) Broad neutralization of wild-type dengue virus isolates following immunization in monkeys with a tetravalent dengue vaccine based on chimeric yellow fever 17D/dengue viruses. Virology 429(2):91–98. https://doi.org/10.1016/j.virol.2012.03.007
Barde PV, Shukla MK, Joshi P, Sahare L, Ukey MJ (2019) Molecular studies on dengue viruses detected in patients from Central India. Indian J Med Microbiol. 37(1):12–18. https://doi.org/10.4103/ijmm.IJMM_18_377
Kolawole OM, Seriki AA, Irekeola AA, Bello KE, Adeyemi OO (2017) Dengue virus and malaria concurrent infection among febrile subjects within Ilorin metropolis, Nigeria. J Med Virol. 89(8):1347–1353. https://doi.org/10.1002/jmv.24788
Mustapha JO, Emeribe AU, Nasir IA (2017) Survey of malaria and anti-dengue virus IgG among febrile HIV-infected patients attending a tertiary hospital in Abuja, Nigeria. HIV AIDS (Auckl) 9:145–151. https://doi.org/10.2147/HIV
Zeng Z, Shi J, Guo X, Mo L, Hu N, Sun J, Wu MN, Zhou HN, Hu YZ (2018) Full-length genome and molecular characterization of dengue virus serotype 2 isolated from an imported patient from Myanmar. Virol J 15(1):131. https://doi.org/10.1186/s12985-018-1043-2
Kyle JL, Harris E (2008) Global spread and persistence of dengue. Annu Rev Microbiol. 62:71–92. https://doi.org/10.1146/annurev.micro.62.081307.163005
Danis-Lozano R, Díaz-González EE, Malo-García IR, Rodríguez MH, Ramos-Castañeda J, Juárez-Palma L, Ramos C, López-Ordóñez T, Mosso-González C, Fernández-Salas I (2019) Vertical transmission of dengue virus in Aedes aegypti and its role in the epidemiological persistence of dengue in Central and Southern Mexico. Trop Med Int Health. 24(11):1311–1319. https://doi.org/10.1111/tmi.13306
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JN, Brownstein JS, Anne GH, Sankoh O, Monica FM, Dylan BG, Thomas J, William WGR, Cameron PS, Thomas WS, Jeremy JF, Simon IH (2013) The global distribution and burden of dengue. Nature 496(7446):504–507. https://doi.org/10.1038/nature12060
Qiu FX, Gubler DJ, Liu JC, Chen QQ (1993) Dengue in China: a clinical review. Bull World Health Organ. 71(3–4):349–359
Wu JY, Lun ZR, James AA, Chen XG (2010) Dengue fever in mainland China. Am J Trop Med Hyg. 83(3):664–671. https://doi.org/10.4269/ajtmh.2010.09-0755
Jiang LY, Jing QL, Liu Y, Cao YM, Su WZ, Biao D, Yang ZC (2017) Molecular characterization and genotype shift of dengue virus strains between 2001 and 2014 in Guangzhou. Epidemiol Infect. 145(4):760–765. https://doi.org/10.1017/S0950268816002429
Zhang HL, Fu SH, Deng Z, Yuan J, Jiang HY, Li MH, Gao XY, Wang JL, Liu YH, Yin ZL, Yang WH, Zhang YZ, Feng Y, Wang HY, Liang GD (2013) An outbreak of imported dengue fever from Myanmar to the border of China, with its viral molecular epidemiological features. Zhonghua Liu Xing Bing Xue Za Zhi. 34(5):428–432
Du JW, Pan XH (2010) Prevalent status and features of dengue fever in China. Zhonghua Liu Xing Bing Xue Za Zhi. 31(12):1429–1433
Hu TS, Zhang HL, Feng Y, Fan JH, Tang T, Liu YH, Zhang L, Yin XX, Chen G, Li HC, Zu J, Li HB, Li YY, Yu J, Zhang FQ, Fan QS (2017) Epidemiological and molecular characteristics of emergent dengue virus in Yunnan Province near the China-Myanmar-Laos border, 2013–2015. BMC Infect Dis. 17(1):331. https://doi.org/10.1186/s12879-017-2401-1
Wang X, Ma D, Huang X, Li L, Li D, Zhao Y, Qiu L, Pan Y, Chen J, Xi J, Shan X, Sun Q (2017) Complete genome analysis of dengue virus type 3 isolated from the 2013 dengue outbreak in Yunnan, China. Virus Res. 238:164–170. https://doi.org/10.1016/j.virusres.2017.06.015
Lin Y, Ma D, Wen S, Zeng F, Hong S, Li L, Li X, Wang X, Ma Z, Pan Y, Chen J, Xi J, Qiu L, Shan X, Sun Q (2019) Molecular characterization of the viral structural gene of the first dengue virus type 1 outbreak in Xishuangbanna: a border area of China, Myanmar, and Laos. Int J Infect Dis. 79:152–161. https://doi.org/10.1016/j.ijid.2018.11.370
Zhao Y, Li L, Ma D, Luo J, Ma Z, Wang X, Pan Y, Chen JY, Xi J, Yang J, Qiu L, Bai C, Jiang L, Shan X, Sun Q (2016) Molecular characterization and viral origin of the 2015 dengue outbreak in Xishuangbanna, Yunnan, China. Sci Rep. 6:34444
Rice CM, Lenches EM, Eddy SR, Shin SJ, Sheets RL, Strauss JH (1985) Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 229(4715):726–733. https://doi.org/10.1126/science.4023707
Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD (2001) Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol Cell. 7(3):593–602. https://doi.org/10.1016/s1097-2765(01)00206-4
Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ (2003) Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 10(11):907–912. https://doi.org/10.1038/nsb990
Chen QQ, Meng YJ, Li Y, Qi XP (2015) Frequency, Duration and intensity of dengue fever epidemic risk in townships in Pearl River Delta and Yunnan in China, 2013. Biomed Environ Sci. 28(5):388–395. https://doi.org/10.3967/bes2015.055
Li Q, Cao W, Ren H, Ji Z, Jiang H (2018) Spatiotemporal responses of dengue fever transmission to the road network in an urban area. Acta Trop. 183:8–13. https://doi.org/10.1016/j.actatropica
Lang AS, Westbye AB, Beatty JT (2017) The distribution, evolution, and roles of gene transfer agents in prokaryotic genetic exchange. Annu Rev Virol. 4(1):87–104. https://doi.org/10.1146/annurev-virology-101416-041624
Penaud-Budloo M, François A, Clément N, Ayuso E (2018) Pharmacology of recombinant adeno-associated virus production. Mol Ther Methods Clin Dev. 8:166–180. https://doi.org/10.1016/j.omtm.2018.01.002
Zhang ZS, Yan YS, Wen YW, Huang HL, Li SQ, He S, Zhang JM (2007) High-level expression of recombinant dengue virus type 2 envelope domain III protein and induction of neutralizing antibodies in BALB/C mice. J Virol Methods. 143(2):125–131. https://doi.org/10.1016/j.jviromet.2007.02.012
Zaneti AB, Yamamoto MM, Sulczewski FB, Almeida BDS, Souza HFS, Ferreira NS, Maeda DLNF, Natiely SNS, Rosa DS, Ferreira LCS, Boscardin SB (2019) Dendritic cell targeting using a DNA vaccine induces specific antibodies and CD4(+) T cells to the dengue virus envelope protein domain III. Front Immunol. 10:59. https://doi.org/10.3389/fimmu.2019.00059
Mota J, Acosta M, Argotte R, Figueroa R, Méndez A, Ramos C (2005) Induction of protective antibodies against dengue virus by tetravalent DNA immunization of mice with domain III of the envelope protein. Vaccine. 23(26):3469–3476. https://doi.org/10.1016/j.vaccine.2004.12.028
Poggianella M, Campos JLS, Chan KR, Tan HC, Bestagno M, Ooi EE, Burrone OR (2015) Dengue E protein domain III-based DNA immunisation induces strong antibody responses to all four viral serotypes. PLoS Negl Trop Dis. 9(7):e0003947. https://doi.org/10.1371/journal.pntd.0003947
Crill WD, Roehrig JT (2001) Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J Virol. 75(16):7769–7773. https://doi.org/10.1128/JVI.75.16.7769-7773.2001
Wahala WM, Silva AM (2011) The human antibody response to dengue virus infection. Viruses 3(12):2374–2395. https://doi.org/10.3390/v3122374
Kawano H, Rostapshov V, Rosen L, Lai CJ (1993) Genetic determinants of dengue type 4 virus neurovirulence for mice. J Virol. 67(11):6567–6575
Acknowledgements
We thank Dr. Penghua Wang of the Department Immunology, School of Medicine, University of Connecticut Health Center, for proofreading and reviewing our manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (31970868), the Foundation of the CAMS (Chinese Academy of Medical Sciences) (Grant no. 2017-I2M-2-006) Initiative for Innovative Medicine (CAMS-I2M) (Grant no. 2016-I2M-1-019), and the Natural Science Foundation of Yunnan Province (Grant no. 2016FA029, 2019ZF004, 2019FD082), the Natural Science Foundation of Yunnan Province Youth Project (grant no. 2019FD082), the Yunnan health training project of high level talents (Grant no. H-2017052).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Ethical approval and consent to participate
Written consent was obtained from each participant or the patient's parents or legal guardians before serum collection in this study. Ethical approval was granted by the Institutional Ethics Committee (Institute of Medical Biology, Chinese Academy of Medical Sciences, and Peking Union Medical College), and the entire study protocol was in accordance with the Declaration of Helsinki for Human Research of 1974 (last modified in 2000).
Consent for publication
All authors reviewed and approved the manuscript for publication.
Additional information
Handling Editor: Tim Skern.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lan, Q., Shu, Y., Li, L. et al. Molecular characterization of structural protein genes of dengue virus serotype 1 epidemic in Yunnan, Southwest China, in 2018. Arch Virol 166, 863–870 (2021). https://doi.org/10.1007/s00705-020-04942-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04942-7