
Abstract
Since 2014, novel non-GII.4 norovirus (NoV) genotypes continue to be reported as the main cause of outbreaks worldwide. In this study, we analyzed the epidemiological and genetic features of NoV outbreaks from July 2014 to June 2018 in Huzhou, China. A total of 450 stool samples collected from 51 AGE outbreaks were tested for NoVs by real-time RT PCR. Partial polymerase and capsid sequences of NoV-positive samples were amplified and sequenced for phylogenetic analysis. NoVs were found to be responsible of 84.3% of AGE outbreaks in Huzhou over the past 5 years. Most NoV outbreaks were reported in the cool months (November-March) and occurred in primary schools and kindergartens. Changes in the diversity of genotypes and the distribution of predominant types were observed in recent years. At least eight genotypes were identified, and 91.9% of the genotyped outbreaks were caused by non-GII.4 strains. The top three circulating genotypes during the study period were GII.2[P16], GII.3[P12], and GII.17[P17]. The predominant NoV genotypes in outbreaks have changed from GII.4 variants to GII.17[P17] in 2014-2015, GII.3[P12] in 2015-2016, and then GII.2[P16] in 2016-2018. Non-GII.4 NoVs play an increasingly important role in outbreaks in Huzhou. Continuous surveillance is needed to monitor the emergence of novel NoV strains and help control NoV outbreaks in the next epidemic season.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Noroviruses (NoVs) are now recognized as the leading cause of viral acute gastroenteritis (AGE) in people of all ages worldwide. Globally, NoVs are estimated to be associated with 50% of all-cause AGE outbreaks and 18% of sporadic cases of AGE [1, 2]. Due to their low infectious dose, prolonged asymptomatic shedding, environmental stability and substantial strain diversity, NoVs are highly infectious and can be easily transmitted from person to person [1]. Outbreaks often occur in semi-closed settings, such as cruise ships, health care settings and schools, that favor person-to-person spread [3].
As a member of the family Caliciviridae, NoV possesses a single-stranded positive-sense RNA genome of 7.7 kb divided into three open reading frames (ORFs) [4]. ORF1 encodes a large polyprotein that is post-translationally cleaved into seven nonstructural proteins, including RNA-dependent RNA polymerase (RdRp). ORF2 and ORF3 encode the major capsid protein VP1and minor capsid protein VP2, respectively [5]. Based on their complete VP1 amino acid sequences, NoVs can be classified genetically into ten different genogroups (GI to GX), but only GI, GII, GIV, GVIII, and GIX (formerly GII.15) are known to infect humans [6,7,8]. GI and GII NoVs are responsible for the majority of disease in humans, whereas GIV viruses are rarely detected. Each genogroup can be subclassified into genotypes. To date, viruses in these ten genogroups can be further divided into 49 confirmed capsid genotypes (9 GI, 27 GII, 3 GIII, 2 GIV, 2 GV, 2 GVI and 1 genotype each for GVII, GVIII, GIX and GX) based on amino acids of the complete VP1 and 60 confirmed P-types (14 GI, 37 GII, 2 GIII, 1 GIV, 2 GV, 2 GVI, 1 GVII and 1 GX) based on partial nucleotide sequences of the RdRp regions [8].
Despite the extensive genetic diversity among NoVs, GII.4 has been the most common genotype that is responsible for the majority of NoV outbreaks and sporadic cases worldwide in the past two decades worldwide [9]. Novel pandemic GII.4 variants have emerged every 2 to 3 years since the mid-1990s, including GII.4 US95/96, Farmington Hills 2002, Hunter 2004, Den Haag 2006, New Orleans 2009, and Sydney 2012.
However, novel non-GII.4 NoV genotypes continue to be reported as the main cause of outbreaks over GII.4 since 2014. In winter 2014–2015, a new GII.17 variant named “Kawasaki” emerged and became the major cause of AGE outbreaks in several countries of Asia [10]. During the 2016-2017 winter season, the uncommon recombinant NoV genotype GII.2[P16] caused a rapid increase of NoV outbreaks in different countries worldwide [11,12,13,14]. This was another event in which non-GII.4 NoV might have become the predominant genotype.
Huzhou is a city in the north of Zhejiang Province, China. It has two districts and three counties, with a total population of 3 million. There are a total of 73 schools in the municipality and its two districts. The Huzhou Center for Disease Control and Prevention is mainly responsible for the detection of norovirus outbreaks at the municipal level and in the two districts. To better understand epidemiologic and genotypic trends of NoV outbreaks in Huzhou since 2014, we analyzed the epidemiological and genetic features of NoV outbreaks from July 2014 to June 2018 in the city of Huzhou.
Materials and methods
AGE outbreak surveillance and specimen collection
In this study, a case of AGE is defined as a person with diarrhea (three or more loose stools) and/or vomiting (two or more times) in a 24-hour period. A confirmed NoV outbreak was defined as the occurrence of three or more epidemiologically linked AGE cases within a period of 3 days with at least two samples testing positive for NoV. In the Huzhou area, district-level CDCs are responsible for conducting outbreak investigations and sample collection. Specimens (stool, anal swab, vomit) from AGE cases were collected and sent to the Huzhou Municipal CDC for NoV detection and further genotyping. A norovirus outbreak surveillance year was defined as starting on 1 July and ending on 30 June of the following year.
NoV detection
Stool and vomit specimens were prepared as 10% (w/v) suspensions in phosphate-buffered saline and then centrifuged for 10 min at 10,000 rpm. Viral RNA was extracted from 200 μl supernatant using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed using genogroup-specific primers and probes for the detection of human GI and GII strains as described previously [15, 16]. Real-time RT-PCR was carried out using a One Step PrimeScript® RT-PCR Kit (DRR064, TaKaRa, Dalian, China). Cycling conditions were as follows: reverse transcription at 42 °C for 30 min, denaturation at 95 °C for 2 min, and 40 cycles of PCR at 94 °C for 5 s and 55 °C for 35 s.
NoV genotyping and phylogenetic analysis
NoV-positive sample was selected for genotyping as described previously [16]. The primer set JV12Y/JV13I was used to amplify a portion of the RdRp gene [17]. The primers G1SKF/G1SKR and G2SKF/G2SK were used to amplify a portion of the VP1 genes of GI and GII strains, respectively [17]. RT-PCR was carried out using a QIAGEN One-Step RT-PCR Kit (QIAGEN, Hilden, Germany) with the following amplification conditions: RT at 50 °C for 30 min and denaturation at 94 °C for 2 min, followed by 35 cycles of 30 s at 94 °C, 30 s at 50 °C, and 1 min at 72 °C, and then a final extension at 72 °C for 10 min. For potential recombinant NoVs strains (i.e. cases where phylogenetic analysis indicated incongruent clustering for the partial sequences of the RdRp and the VP1 genes), sequences covering the overlap between ORF1 and ORF2 were further amplified by RT-PCR using the primers JV12 and G1SKR/G2SKR. The PCR products were sent to TaKaRa Biotechnology (Dalian, China) for further purification and sequencing. Genotypes were initially determined using the online Norovirus Typing Tool (https://www.rivm.nl/mpf/typingtool/norovirus/) [18]. Phylogenetic analysis was performed using MEGA software (version 6.06) [19]. The phylogenetic tree was constructed using the neighbor-joining method and validated by 1000 bootstrap replicates.
Accession numbers
Sequences obtained in this study were deposited in the GenBank database under accession numbers KY344405 to KY344414, KY344277 to KY344297, KY344329 to KY344333, KY344342 to KY344354, MG763356 to MG763364, MG763378 to MG763382, MG763385 to MG763401, MF073228 to MF073230, MN210012, MN210018, MN210023, MN186383, MG739461 to MG739462, MG739464 to MG739465, MG739470 to MG739471, MG739296 to MG739297, MG739300 to MG739301, MG739309 to MG739310, and MG739306.
Results
Epidemiological features of NoV outbreaks in Huzhou, China, from 2014 to 2018
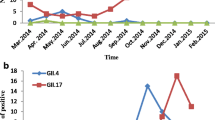
From July 2014 to June 2018, 199 of 450 specimens collected from 51 AGE outbreaks were positive for NoV by real-time RT-PCR. During this period, NoVs were involved in 43 (84.3%) of the AGE outbreaks, including 8 outbreaks detected from 2014 to 2015, 9 outbreaks detected from 2015 to 2016, 18 outbreaks detected from 2016 to 2017, and 8 outbreaks detected from 2017 to 2018 (Table 1). Of the 43 NoV outbreaks, 40 (93.0%) were caused by GII NoV strains, 2 (4.7%) were caused by GI NoV strains, and 1 (2.3%) was caused by a combination of GI and GII NoV strains. In this outbreak caused by mixed infections with GI and GII, NoVs were detected in five stool samples from patients, three of which were positive for both GI and GII, and two of which were positive for GII. As for seasonal distribution, NoV outbreaks occurred throughout the year, except in summer. Most (30/43, 69.8%) of the outbreaks occurred from November to the following March, while no outbreaks were identified from June to August (Fig. 1A). The majority of outbreaks (88.4%, 38/43) occurred in school settings, mainly in primary schools (n = 17, 39.5%) and kindergartens (n = 15, 34.9%), followed by middle schools (n = 6, 14.0%). Other outbreak settings include hospitals, communities, and tour groups, accounting for 4.7% (n = 2), 4.7% (n = 2), and 2.3% (n = 1), respectively, of all NoV outbreaks.

Monthly genotype distribution of NoV outbreaks in Huzhou, China, between July 2014 and June 2018
Norovirus genotyping
Of the 199 NoV-positive samples confirmed by real-time RT-PCR, 86 samples from 37 outbreaks were successfully sequenced and genotyped by RT-PCR. Eight genotypes based on both RdRp and capsid genes were identified throughout the study period: GI.6[P11] (formerly GI.6[Pb]), GI.5[P4], GII.2[P2], GII.6[P7], GII.4 Sydney[P31] (formerly GII.4 Sydney[Pe]), GII.17[P17], GII.3[P12] and GII.2[P16] (Table 1). 91.9% (34/37) of the genotyped outbreaks were caused by non-GII.4 strains. GII.2[P16], GII.3[P12] and GII.17[P17] were the top three circulating genotypes during the study period, accounting for at least 32.6% (14/43), 20.9% (9/43), and 11.6 (5/43) of the NoV-positive outbreaks, respectively. The previous predominant genotype GII.4 Sydney2012 was only responsible for 7.0% (3/43) of the outbreaks, ranking fourth. The predominant genotypes of NoV during our study period varied according to surveillance year. Overall, GII.17[P17] genotypes predominated in outbreaks during 2014–2015, accounting for 62.5% (5/8) of the outbreaks. GII.3[P12] genotypes predominated in 2015–2016, causing 55.6% (5/9) of the outbreaks. GII.2[P16] was the predominant genotype both in 2016-2017 and 2017-2018, responsible for 50.0% (9/18) and 62.5% (5/8) of the NoV outbreaks, respectively.
The genotype distribution of NoV outbreaks in each month is shown in Fig. 1. GII.17 was first detected in the outbreak in November 2014 and soon became the only genotype detected in the first half of 2015. However, no outbreak of GII.17 infection occurred after May 2015, except for one outbreak caused by a mixed infection of GI.5 [P4] and GII.17 [P17] in March 2018. GII.3[P12] subsequently became the most frequently detected genotype in outbreaks between July 2015 and December 2016 (7 of 13 NoV outbreaks, 53.8%). NoV GII.2[P16] was first detected in an outbreak in February 2017, and it went on to replace GII.3[P12] as the predominant genotype from February 2017 to June 2018. With the emergence of GII.2[P16], we observed a sharp increase in the number of NoV outbreaks during February and May 2017. Compared with five outbreaks caused by GII.17 in 2014-2015, GII.2[P16] caused nine outbreaks in 2016-2017.
We also compared the settings of NoV outbreaks caused by GII.17[P17], GII.3[P12], and GII.2[P16] (Table 2). Most GII.17[P17] outbreaks were reported in primary schools (80%, 4/5), whereas GII.2[P16] outbreaks occurred in all school types. All of the GII.3[P12] outbreaks occurred in kindergartens and primary schools, affecting children ≤ 12 years of age.
Phylogenetic analysis
Based on the online genotyping results, we selected one representative strain with the same genotype from each outbreak for phylogenetic analysis. In total, 38 strains from 37 outbreaks were subjected to phylogenetic analysis based on both partial RdRp and VP1 gene sequences (Fig. 2 and Fig. 3). Genotype inconsistency between the RdRp and VP1 regions was found in 30 strains (indicated by red triangles). Of the genotyped outbreaks, 81.1% (30/37) were caused by recombinant strains, including GI.6[P11], GI.5[P4], GII.6[P7], GII.4 Sydney[P31], GII.3[P12], and GII.2[P16]. Phylogenetic analysis showed that GII.17 strains collected during our study period all clustered together and belonged to the novel GII.P17 Kawasaki cluster. GII.2[P16] strains detected in Huzhou outbreaks during 2017 -2018 were clustered with GII.2[P16] strains detected by the end of 2016 in China, Germany, USA, and France in both the RdRp and capsid region and formed a relatively independent subcluster (designated as the 2016 subcluster).

Phylogenetic analysis based on partial RdRp gene (A) and capsid gene (B) sequences of GI NoVs. NoV strains identified in Huzhou are designated by location, year, and sample number (indicated by triangles). Recombinants identified in this study are highlighted in red

Phylogenetic analysis based on partial RdRp gene (A) and capsid gene (B) sequences of GII NoVs. NoV strains identified in Huzhou are designated by location, year, and sample number (indicated by triangles). Recombinants identified in this study are highlighted in red
Discussion
In this study, a systematic investigation of the molecular epidemiology of NoV outbreaks was conducted in Huzhou from July 2014 to June 2018. NoV was detected in 43 (84.3%) of the AGE outbreaks. GII NoV strains were largely predominant and responsible for 93.0% of laboratory-confirmed NoV outbreaks. Similar to other cities in the Northern Hemisphere [20,21,22], NoV outbreaks in Huzhou exhibited a clear seasonality, with a peak in the cool months (November-March) and a trough in the summer months. The majority of outbreaks (88.4%, 38/43) occurred in school settings, mainly in primary schools and kindergartens. A higher percentage of NoV outbreaks among schools was also observed in other regions of China [23, 24]. From January 2014 to December 2017, a total of 616 NoV outbreaks were reported to the National Public Health Emergency Event Surveillance System (PHEESS) in China. Among these outbreaks, 93% occurred in school settings, including 39% in primary schools, 22% in childcare facilities, and 20% in secondary schools. However, this differs from the United States and Europe, where long-term care facilities (e.g., nursing homes) were reported as the most common settings [25,26,27]. In China, young students stay in crowded, enclosed classrooms (with an average of 30–50 students per classroom), where the viruses are highly transmissible by close contact between students, particularly among young students with lower levels of hand hygiene [28]. In addition, the high rate of NoV outbreaks in schools may also be due to the surveillance bias among different institutions. The Chinese Ministry of Education requires schools to screen students attending kindergarten, primary, and junior high schools every morning to check for fever, vomiting, or diarrhea [24]. Therefore, the reporting of infectious diseases including NoV outbreaks is better in these school settings.
In the Huzhou area, GII.4 has been the only predominant genotype in outbreaks and sporadic cases of AGE since 2008 [16, 29]. From 2008 to 2012, 80% of typed NoV outbreaks were caused by GII.4 variants, including GII.4 2006b, New Orleans 2009, and Sydney2012. All of the sequenced GII NoVs belonged to the GII.4 genotype except one strain, which had a GII.b polymerase gene. From July 2014 to June 2018, NoV strains associated with the outbreaks in Huzhou exhibited important changes in the diversity of genotypes and the distribution of predominant types as compared to previous study data. At least eight genotypes were identified during the study period, including GI.6[P11], GI.5[P4], GII.2[P2], GII.6[P7], GII.4 Sydney[P31], GII.17[P17], GII.3[P12], and GII.2[P16]. Of the genotyped outbreaks, 91.9% were caused by non-GII.4 strains. The three most common genotypes were GII.2[P16], GII.3[P12], and GII.17[P17], while the previous predominant genotype, GII.4 Sydney2012, only ranked fourth. Similar results were observed in other cities of China. In Beijing, the capital of China, 92.3% of the laboratory-confirmed NoV outbreaks occurred in kindergartens and elementary schools during 2014-2017, and the most commonly identified strains in outbreaks were GII.2[P16], GII.17[P17], and GII.3[P12] [30]. In Jinan, a city in eastern China, seven genotypes were prevalent in outbreaks of AGE from 2015 to 2017, while no GII.4 was identified [31]. From January 2015 to December 2018, the Jiangsu Provincial Center for Disease Control and Prevention reported a total of 213 NoV outbreaks, most of which occurred in kindergartens and elementary schools. At least 83.6% of outbreaks were caused by GII recombinant strains, of which GII.4 Sydney [P31] accounted for only 0.5% [32]. Based on the above results, we speculate that non-GII.4 NoV plays an increasingly important role in the outbreaks of AGE in China.
Globally, new GII.2[P16] strains have a wider epidemic range than GII.17[P17] strains. A novel GII.17 NoV strain first emerged in Asia in September 2014 and soon became the predominant genotype in some areas in Asia during the 2014–2015 winter season, but GII.17 appears to have had limited prevalence in other regions outside Asia, including Europe, Canada, and the United States [10, 33, 34]. By contrast, in late 2016, uncommon recombinant GII.2[P16] strains emerged as the major cause of AGE outbreaks in different countries and caused an increase in NoV activity worldwide [11,12,13,14]. In Huzhou, GII.17 was first detected in an outbreak in November 2014 and became the predominant strain in the first half of 2015, but after that, the prevalence of GII.17 declined rapidly. After GII.2[P16] emerged in December 2016, it went on to become the main genotype detected in outbreaks until June 2018. Compared with five outbreaks caused by GII.17 in 2014-2015, GII.2[P16] caused nine outbreaks in 2016-2017. GII.17[P17] outbreaks were concentrated in primary schools in Huzhou, while GII.2[P16] outbreaks had a larger epidemic range, occurring in all school types. The above results suggest that GII.2[P16] strains have superior epidemiological adaptability in Huzhou to GII.17[P17] strains. A similar finding was reported in Beijing, where the GII.2[P16] strains caused a larger number of outbreaks and had a broader epidemic range than GII.17[P17]. The factors that limit the spread of GII.17 are unknown. Such restrictions could be due to differences in the existing immunity of the population or differences between populations in the expression of NoV receptors [10, 35].
Unlike GII.17[P17] and GII.2[P16], which emerged as “novel” genotypes and subsequently became the dominant strain during the epidemic season, GII.3[P12] has been prevalent as one of the major recombinant types detected in children in Asian countries, including South Korea, Japan, and China, for several years [36,37,38,39,40]. Similarly, in our study, all GII.3[P12] outbreaks occurred in kindergartens and elementary schools, mainly affecting children under 12 years of age. Previous studies indicated that GII.3[P12] has been detected in sporadic cases of AGE in Huzhou since 2013, but it has never been the dominant type. However, in our study, GII.3[P12] surpassed GII.17 in the second half of 2015 and became the main genotype detected in NoV outbreaks in the 2015-2016 epidemic season. Similar findings were reported in Beijing, where GII.3[P12] was the major genotype responsible for outbreaks during 2015–2016 [30]. After so many years since its emergence, why did GII.12-GII.3 show a dominant position in outbreaks in the 2015-2016 season? The reason for this is unknown. Further analysis using long sequences covering the complete RdRp and VP1 region are required to explore the evolutionary dynamics of GII.3[P12] strains.
Our study has several limitations. First, genotyping was only successful for 86 (43.2%) of the NoV strains detected. Second, the number of outbreaks we investigated was relatively small. Due to surveillance bias, outbreak identification and reporting may differ across settings, and therefore, the most prevalent genotype in settings (especially for the settings outside the school system) with lower reporting rates may have been underrepresented in our analysis.
In conclusion, we analyzed the epidemiological and genetic features of NoV outbreaks from July 2014 to June 2018 in Huzhou. Most NoV outbreaks were reported in the cool months (November-March) and occurred in primary schools and kindergartens. Compared to previous study data, differences in the diversity of genotypes and the distribution of predominant types were observed. The predominant NoV genotypes in outbreaks changed from GII.4 variants to GII.17[P17] in 2014-2015, GII.3[P12] in 2015-2016, and then GII.2[P16] in 2016-2018. Our findings demonstrate that non-GII.4 NoVs play an increasingly important role in the outbreaks in Huzhou. Continuous surveillance is needed to monitor the emergence of novel NoV strains and help control NoV outbreaks in the next epidemic season.
References
Patel MM, Hall AJ, Vinje J, Parashar UD (2009) Noroviruses: a comprehensive review. J Clin Virol 44(1):1–8. https://doi.org/10.1016/j.jcv.2008.10.009
Ahmed SM, Hall AJ, Robinson AE, Verhoef L, Premkumar P, Parashar UD, Koopmans M, Lopman BA (2014) Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. Lancet Infect Dis 14(8):725–730. https://doi.org/10.1016/s1473-3099(14)70767-4
Glass RI, Parashar UD, Estes MK (2009) Norovirus gastroenteritis. N Engl J Med 361(18):1776–1785. https://doi.org/10.1056/NEJMra0804575
Green KY, Ando T, Balayan MS, Berke T, Clarke IN, Estes MK, Matson DO, Nakata S, Neill JD, Studdert MJ, Thiel HJ (2000) Taxonomy of the caliciviruses. J Infect Dis 181(Suppl 2):S322–S330. https://doi.org/10.1086/315591
Thorne LG, Goodfellow IG (2014) Norovirus gene expression and replication. J Gen Virol 95(Pt 2):278–291. https://doi.org/10.1099/vir.0.059634-0
Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS (2006) Norovirus classification and proposed strain nomenclature. Virology 346(2):312–323. https://doi.org/10.1016/j.virol.2005.11.015
Vinje J (2015) Advances in laboratory methods for detection and typing of norovirus. J Clin Microbiol 53(2):373–381. https://doi.org/10.1128/jcm.01535-14
Chhabra P, de Graaf M, Parra GI, Chan MC, Green K, Martella V, Wang Q, White PA, Katayama K, Vennema H, Koopmans MPG, Vinje J (2019) Updated classification of norovirus genogroups and genotypes. J Gen Virol 100(10):1393–1406. https://doi.org/10.1099/jgv.0.001318
Bok K, Abente EJ, Realpe-Quintero M, Mitra T, Sosnovtsev SV, Kapikian AZ, Green KY (2009) Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J Virol 83(22):11890–11901. https://doi.org/10.1128/jvi.00864-09
de Graaf M, van Beek J, Vennema H, Podkolzin AT, Hewitt J, Bucardo F, Templeton K, Mans J, Nordgren J, Reuter G, Lynch M, Rasmussen LD, Iritani N, Chan MC, Martella V, Ambert-Balay K, Vinje J, White PA, Koopmans MP (2015) Emergence of a novel GII.17 norovirus—end of the GII.4 era? Euro Surveill 20 (26)
Ao Y, Wang J, Ling H, He Y, Dong X, Wang X, Peng J, Zhang H, Jin M, Duan Z (2017) Norovirus GII.P16/GII.2-associated gastroenteritis, China, 2016. Emerg Infect Dis 23(7):1172–1175. https://doi.org/10.3201/eid2307.170034
Bidalot M, Thery L, Kaplon J, De Rougemont A, Ambert-Balay K (2017) Emergence of new recombinant noroviruses GII.p16-GII.4 and GII.p16-GII.2, France, winter 2016 to 2017. Euro Surveill 22(15). https://doi.org/10.2807/1560-7917.es.2017.22.15.30508
Niendorf S, Jacobsen S, Faber M, Eis-Hubinger AM, Hofmann J, Zimmermann O, Hohne M, Bock CT (2017) Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Euro Surveill 22(4). https://doi.org/10.2807/1560-7917.es.2017.22.4.30447
Nagasawa K, Matsushima Y, Motoya T, Mizukoshi F, Ueki Y, Sakon N, Murakami K, Shimizu T, Okabe N, Nagata N, Shirabe K, Shinomiya H, Suzuki W, Kuroda M, Sekizuka T, Ryo A, Fujita K, Oishi K, Katayama K, Kimura H (2018) Phylogeny and immunoreactivity of norovirus GII.P16-GII.2, Japan, Winter 2016–17. Emerg Infect Dis 24 (1):144–148. https://doi.org/10.3201/eid2401.170284
Jothikumar N, Lowther JA, Henshilwood K, Lees DN, Hill VR, Vinje J (2005) Rapid and sensitive detection of noroviruses by using TaqMan-based one-step reverse transcription-PCR assays and application to naturally contaminated shellfish samples. Appl Environ Microbiol 71(4):1870–1875. https://doi.org/10.1128/aem.71.4.1870-1875.2005
Ji L, Wu X, Yao W, Chen L, Xu D, Shen Y, Shen J, Han J (2013) Rapid emergence of novel GII.4 sub-lineages noroviruses associated with outbreaks in Huzhou, China, 2008–2012. PLoS One 8(12):e82627. https://doi.org/10.1371/journal.pone.0082627
Vennema H, de Bruin E, Koopmans M (2002) Rational optimization of generic primers used for Norwalk-like virus detection by reverse transcriptase polymerase chain reaction. J Clin Virol 25(2):233–235
Kroneman A, Vega E, Vennema H, Vinje J, White PA, Hansman G, Green K, Martella V, Katayama K, Koopmans M (2013) Proposal for a unified norovirus nomenclature and genotyping. Arch Virol 158(10):2059–2068. https://doi.org/10.1007/s00705-013-1708-5
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729. https://doi.org/10.1093/molbev/mst197
Ahmed SM, Lopman BA, Levy K (2013) A systematic review and meta-analysis of the global seasonality of norovirus. PLoS One 8(10):e75922. https://doi.org/10.1371/journal.pone.0075922
Thongprachum A, Khamrin P, Maneekarn N, Hayakawa S, Ushijima H (2016) Epidemiology of gastroenteritis viruses in Japan: Prevalence, seasonality, and outbreak. J Med Virol 88(4):551–570. https://doi.org/10.1002/jmv.24387
Cho HW, Chu C (2015) Norovirus outbreaks occurred in different settings in the Republic of Korea. Osong Public Health Res Perspect 6(5):281–282. https://doi.org/10.1016/j.phrp.2015.11.001
Qin SW, Chan TC, Cai J, Zhao N, Miao ZP, Chen YJ, Liu SL (2017) Genotypic and epidemiological trends of acute gastroenteritis associated with noroviruses in China from 2006 to 2016. Int J Environ Res Public Health 14(11). https://doi.org/10.3390/ijerph14111341
Lian Y, Wu S, Luo L, Lv B, Liao Q, Li Z, Rainey JJ, Hall AJ, Ran L (2019) Epidemiology of norovirus outbreaks reported to the public health emergency event surveillance system, China, 2014(—)2017. Viruses 11(4). https://doi.org/10.3390/v11040342
Vega E, Barclay L, Gregoricus N, Shirley SH, Lee D, Vinje J (2014) Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J Clin Microbiol 52(1):147–155. https://doi.org/10.1128/JCM.02680-13
Friesema IH, Vennema H, Heijne JC, de Jager CM, Morroy G, van den Kerkhof JH, de Coster EJ, Wolters BA, ter Waarbeek HL, Fanoy EB, Teunis PF, van der Linde R, van Duynhoven YT (2009) Norovirus outbreaks in nursing homes: the evaluation of infection control measures. Epidemiol Infect 137(12):1722–1733. https://doi.org/10.1017/S095026880900274X
Barret AS, Jourdan-da Silva N, Ambert-Balay K, Delmas G, Bone A, Thiolet JM, Vaillant V (2014) Surveillance for outbreaks of gastroenteritis in elderly long-term care facilities in France, November 2010 to May 2012. Euro Surveill 19 (29). https://doi.org/10.2807/1560-7917.es2014.19.29.20859
Li J, Gao X, Ye YL, Wan T, Zang H, Mo PH, Song CL (2018) An acute gastroenteritis outbreak associated with person-to-person transmission in a primary school in Shanghai: first report of a GI.5 norovirus outbreak in China. BMC Infect Dis 18(1):316. https://doi.org/10.1186/s12879-018-3224-4
Wu X, Han J, Chen L, Xu D, Shen Y, Zha Y, Zhu X, Ji L (2015) Prevalence and genetic diversity of noroviruses in adults with acute gastroenteritis in Huzhou, China, 2013-2014. Arch Virol 160(7):1705–1713. https://doi.org/10.1007/s00705-015-2440-0
Gao Z, Liu B, Yan H, Li W, Jia L, Tian Y, Chen Y, Wang Q, Pang X (2019) Norovirus outbreaks in Beijing, China, from 2014 to 2017. J Infect 79(2):159–166. https://doi.org/10.1016/j.jinf.2019.05.019
Liu L, Guan H, Zhang Y, Wang C, Yang G, Ruan S, Zhao H, Han X (2018) The prevalence of non-GII.4 norovirus genotypes in acute gastroenteritis outbreaks in Jinan, China. PLoS One 13(12):e0209245. https://doi.org/10.1371/journal.pone.0209245
Fu J, Bao C, Huo X, Hu J, Shi C, Lin Q, Zhang J, Ai J, Xing Z (2019) Increasing recombinant strains emerged in norovirus outbreaks in Jiangsu, China: 2015-2018. Sci Rep 9(1):20012. https://doi.org/10.1038/s41598-019-56544-2
Hasing ME, Lee BE, Qiu Y, Xia M, Pabbaraju K, Wong A, Tipples G, Jiang X, Pang XL (2019) Changes in norovirus genotype diversity in gastroenteritis outbreaks in Alberta, Canada: 2012-2018. BMC Infect Dis 19(1):177. https://doi.org/10.1186/s12879-019-3792-y
Parra GI (2014) Green KY (2015) Genome of Emerging Norovirus GII.17, United States. Emerg Infect Dis 21(8):1477–1479. https://doi.org/10.3201/eid2108.150652
Siebenga JJ, Vennema H, Zheng DP, Vinje J, Lee BE, Pang XL, Ho EC, Lim W, Choudekar A, Broor S, Halperin T, Rasool NB, Hewitt J, Greening GE, Jin M, Duan ZJ, Lucero Y, O’Ryan M, Hoehne M, Schreier E, Ratcliff RM, White PA, Iritani N, Reuter G, Koopmans M (2009) Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001-2007. J Infect Dis 200(5):802–812. https://doi.org/10.1086/605127
Guo L, Song J, Xu X, Ren L, Li J, Zhou H, Wang M, Qu J, Wang J, Hung T (2009) Genetic analysis of norovirus in children affected with acute gastroenteritis in Beijing, 2004-2007. J Clin Virol 44(1):94–98. https://doi.org/10.1016/j.jcv.2008.10.002
Phan TG, Kuroiwa T, Kaneshi K, Ueda Y, Nakaya S, Nishimura S, Yamamoto A, Sugita K, Nishimura T, Yagyu F, Okitsu S, Muller WE, Maneekarn N, Ushijima H (2006) Changing distribution of norovirus genotypes and genetic analysis of recombinant GIIb among infants and children with diarrhea in Japan. J Med Virol 78(7):971–978. https://doi.org/10.1002/jmv.20649
Han TH, Kim CH, Chung JY, Park SH, Hwang ES (2011) Emergence of norovirus GII-4/2008 variant and recombinant strains in Seoul, Korea. Arch Virol 156(2):323–329. https://doi.org/10.1007/s00705-010-0844-4
Hoa Tran TN, Trainor E, Nakagomi T, Cunliffe NA, Nakagomi O (2013) Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: global distribution of genogroups, genotypes and GII.4 variants. J Clin Virol 56(3):185–193. https://doi.org/10.1016/j.jcv.2012.11.011
Lu QB, Huang DD, Zhao J, Wang HY, Zhang XA, Xu HM, Qu F, Liu W, Cao WC (2015) An increasing prevalence of recombinant GII norovirus in pediatric patients with diarrhea during 2010-2013 in China. Infect Genet Evol 31:48–52. https://doi.org/10.1016/j.meegid.2015.01.008
Acknowledgments
This study was supported by grants from the Zhejiang Medical Science and Technology Program (Grant no. 2018RC015) and the Natural Science Foundation of the Huzhou Science and Technology Bureau (Grant no. 2017YZ04).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This study was approved by the human research ethics committee of the Huzhou Center for Disease Control and Prevention (CDC). Informed consent for the use of the stool samples was obtained from the patients or their guardians.
Additional information
Handling Editor: Reimar Johne.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, L., Xu, D., Wu, X. et al. An increasing prevalence of non-GII.4 norovirus genotypes in acute gastroenteritis outbreaks in Huzhou, China, 2014-2018. Arch Virol 165, 1121–1128 (2020). https://doi.org/10.1007/s00705-020-04599-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04599-2