
Abstract
A novel lymphocystivirus causing typical signs of lymphocystis virus disease in whitemouth croaker (Micropogonias furnieri) on the coast of Uruguay was detected and described recently. Based on genetic analysis of some partially sequenced core genes, the virus seemed to differ from previously described members of the genus Lymphocystivirus. In this study, using next-generation sequencing, the whole genome of this virus was sequenced and analysed. The complete genome was found to be 211,086 bp in size, containing 148 predicted protein-coding regions, including the 26 core genes that seem to have a homologue in every iridovirus genome sequenced to date. Considering the current species demarcation criteria for the family Iridoviridae (genome organization, G+C content, amino acid sequence similarity, and phylogenetic relatedness of the core genes), the establishment of a novel species (“Lymphocystis disease virus 4”) in the genus Lymphocystivirus is suggested.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Lymphocystis disease viruses (LCDVs) belong to the genus Lymphocystivirus, family Iridoviridae. LCDVs are large (200-230 nm) dsDNA viruses. Their genome, which ranges in size between 108 and 208 kilobasepairs (kbp), is circularly permuted, terminally redundant, and heavily methylated at cytosines in CpG sequences, and these viruses typically have a low G+C content (27-33%) [4, 7]. LCDVs infect a wide range of fish species (over 150). A common disease sign is the development of macroscopic nodules (0.3-2.0 mm) located on the body surface and fins [1]. The genus Lymphocystivirus currently includes three virus species accepted by the International Committee on Taxonomy of Viruses (ICTV): Lymphocystis disease virus 1, 2 and 3. The complete genome sequence of the LCDV-1 was published in the 1990s [14], and this virus has been reported in flatfishes (family Pleuronectidae) [3]. LCDV-2 was isolated also from a flatfish species, namely flounder (Paralichthys olivaceus) [15], while LCDV-3 was found in gilthead sea bream (Sparus aurata) [11].
A few years ago, an outbreak of lymphocystis disease (LCD) was detected in wild and cultured populations of whitemouth croaker (Micropogonias furnieri) on the coast of Uruguay. Molecular analysis targeting some of the iridoviral core genes showed the presence of the DNA of an unknown LCDV in all specimens showing external signs of LCD. Phylogenetic analysis based on the concatenated sequences of six partially sequenced core genes suggested that the virus belongs to the genus Lymphocystivirus. However, the sequences of the whitemouth croaker LCDV (LCDV-WC) differed markedly from those of members of the three accepted species in this genus, putatively representing a fourth viral species in the genus. In the present study, using next-generation sequencing, the whole genome of this virus was sequenced and analysed.
Diseased fish were collected on the coast of Uruguay [13]. Samples from the lesions were conserved in ethanol for molecular studies. Total DNA was extracted from the samples using a DNeasy Blood and Tissue Kit (QIAGEN, Germany). A short-insert DNA library was prepared and sequenced using the Illumina HiSeqTM 2000 platform (Illumina, USA). A total of 16,834,698 paired-end reads were generated (Q20% = 96.33), with the average length of 66 bp; CLC Genomics Workbench 12.0 (CLC bio, Denmark) and Geneious 11.1.5 (Biomatters Ltd., New Zealand) were used for genome assembly (average read depth = 2916). There were two short gaps between the contigs. These gaps were closed and some further sequence ambiguities were resolved using Sanger sequencing with PCR primers designed based on the flanking regions of sequences. FGENESV was used for prediction of open reading frames (ORFs) (Softberry, Inc., USA). The complete genome sequence of the novel LCDV was deposited in the GenBank database under the accession number MN803438. The genome sequence was compared to those of LCDV-1, -2 and -3 using LASTZ 1.02.00 in Geneious 11.1.5 (Biomatters Ltd., New Zealand). In LASTZ, default settings were used: the high-scoring segment pairs—homologous stretches—were scored using the HOXD70 substitution scores [2], and the lower score threshold was 3000. The deduced amino acid sequences of the proteins encoded by the 26 core genes (Supplementary Table 1) were concatenated, and this sequence was used for phylogenetic analysis. For tree inference, a multiple alignment of the concatenates was made using Mafft v7 [10] with default parameters, and the alignment was edited manually. Evolutionary model selection was done using ModelTest-NG v0.1.5 [5], and the LG+I+G model had the highest probability. The phylogenetic calculation was performed using RAxML-NG v0.9.0 [8], the robustness of the tree was analyzed using a non-parametric bootstrap calculation with 1,000 repeats. The phylogenetic tree was visualized using MEGA 7 [9], and bootstrap values are given as percentages. A pairwise sequence identity analysis was also conducted on the same concatenate of the four LCDVs, using SDT 1.2 [12].
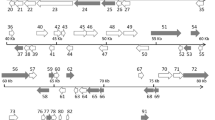
The complete genome of the LCDV-WC was found to be 211,086 bp in size. The G+C content of the whole genome was 26.0%. Comparison of the genome sequence of LCDV-WC to those of previously described LCDVs showed that the genome size of LCDV-WC is the longest, and its G+C content is the lowest (LCDV-1, 29.1%; LCDV-2, 27.2%; LCDV-3, 33.0%). The genome organization of the LCDV-WC shows similarity to that of LCDV-2 and -3, but major rearrangements are also observable (Fig. 1). The LCDV-WC genome was predicted to contain 148 ORFs. The majority of the ORFs (102) showed clear homology to the genes of all three other LCDVs. The 26 core genes, which are conserved in all sequenced iridoviruses, were also identified in the genome. Nine ORFs lacked similarity to any known viral gene. The rest of the putative genes showed homology to genes of only one or two of the LCDVs. The protein product concatenate of the core genes showed 67.1–85.1% amino acid sequence identity to its LCDV counterparts (Fig. 2). The phylogenetic tree reconstruction clearly illustrates that LCDV-WC clusters with members of the genus Lymphocystivirus and shows a clear separation of LCDV-WC from the other LCDVs (Fig. 3).

Comparison of the whitemouth croaker lymphocystis disease virus (LCDV-WC) genome sequence to those of LCDV1-3. The genome sequence of LCDV-WC is on the abscissa, and the genome sequences of LCDV1, LCDV2, and LCDV3 are on the ordinate in separate sections. Blue bars represent homologous high-scoring segment pairs in a codirectional orientation, whereas red bars represent reversed pairs. LCDV, lymphocystis disease virus

Pairwise sequence identity analysis of 26 core gene amino acid sequence concatenates of LCDV1-3 and whitemouth croaker lymphocystis disease virus. LCDV, lymphocystis disease virus; LCDV-WC, whitemouth croaker lymphocystis disease virus. Pairwise sequence identity percentages are represented using different colours. Cutoff values: 85% and 95%. Blue squares represent similarity below 85%; green squares, 85-95%; red squares, ≥95%

Phylogenetic analysis of iridoviruses. Maximum-likelihood analysis of 26 core gene amino acid sequence concatenates (8178 aa). The tree is rooted at the midpoint. Bootstrap values are given as percentages. ADRV, Andrias davidianus ranavirus; AMIV, Anopheles minimus iridovirus; ATV, Ambystoma tigrinum virus; BIV, Bohle iridovirus; CH, Testudo hermanni ranavirus; CMTV, common midwife toad virus; CoIV, cod iridovirus; DFV, doctor fish virus; ECV, European catfish virus; EHNV, epizootic haematopoietic necrosis virus; ESV, European sheatfish virus; FV, frog virus; GGRV, German gecko ranavirus; GIV, grouper iridovirus; GSIV, giant seaperch iridovirus; GV, guppy virus; IIV, invertebrate iridescent virus; ISKNV, infectious skin and kidney necrosis virus; LCDV, lymphocystis disease virus; LMBV, largemouth bass virus; LYCIV, lemon yellow croaker iridovirus; OSGIV, orange-spotted grouper iridovirus; PPIV, pike perch iridovirus; RBIV, rock bream iridovirus; RGV, Rana grylio virus; Rmax, Rana maxima virus; RSIV, red seabream iridovirus; SDDV, scale drop disease virus; SERV, short-finned eel ranavirus; SGIV, Singapore grouper iridovirus; SSME, spotted salamander Maine virus; STIV, soft-shelled turtle iridovirus; TFV, tiger frog virus; ToRV, tortoise ranavirus; TRBIV, turbot reddish body iridovirus
There are 26 well-conserved core genes in the genomes of all known and completely sequenced iridoviruses, the products of which are associated with a variety of viral activities, including DNA metabolism, transcriptional regulation, protein modification, and viral structure [6]. According to the current species demarcation criteria for the members of the family Iridoviridae (https://talk.ictvonline.org/files/ictv_official_taxonomy_updates_since_the_8th_report/m/animal-dna-viruses-and-retroviruses/8054), viruses sharing 95% or greater amino acid sequence identity in the predicted products of their core genes should be considered members of the same species. Moreover, members of the same species have to have a similar genome size and G+C content, and they should show phylogenetic relatedness and a collinear gene arrangement. The analysis of the complete genome sequence of LCDV-WC confirmed that this virus is a member of a distinct species in the genus Lymphocystivirus, as was suspected from partial sequence information from a previous study [13]. This demonstrates that complete genome sequences may not be necessary for establishing a novel species. The authors propose that the establishment of the new species “Lymphocystis disease virus 4” should be considered for approval by the ICTV.
References
Anders K (1989) Lymphocystis disease of fishes. In: Ahne W, Kurstak E (eds) Viruses of lower vertebrates. Springer, Berlin, pp 140–160
Chiaromonte F, Yap VB, Miller W (2002) Scoring pairwise genomic sequence alignments. Pac Symp Biocomput. https://doi.org/10.1142/9789812799623_0012
Darai G, Anders K, Koch H, Delius H, Gelderblom H, Samalecos C, Flugel R (1983) Analyisis of the genome of fish lymphocystis disease virus isolated directly from epidermal tumors of pleuronectes. Virology 126:466–479
Darai G, Delius H, Koch H, Flugel R (1983) The genome of fish lymphocystis disease virus (FLDV) is circularly permuted and terminally redundant. Zentralblatt Fur Bakteriologie Mikrobiologie Und Hygiene Ser A Med Microbiol Infect Diseases Virol Parasitol 254:155
Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T (2020) ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol 37(1):291–294. https://doi.org/10.1093/molbev/msz189
Eaton HE, Metcalf J, Penny E, Tcherepanov V, Upton C, Brunetti CR (2007) Comparative genomic analysis of the family Iridoviridae: re-annotating and defining the core set of iridovirus genes. Virol J 4:11
Jancovich J, Chinchar V, Hyatt A, Miyazaki T, Williams T, Zhang Q (2011) Family Iridoviridae. In: King A, Adams M, Carstens E, Lefkowitz E (eds) Virus taxonomy: classification and nomenclature of viruses ninth report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego, pp 193–210
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kuraku S, Zmasek CM, Nishimura O, Katoh K (2013) aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res 41:W22–28
López-Bueno A, Mavian C, Labella AM, Castro D, Borrego JJ, Alcami A, Alejo A (2016) Concurrence of iridovirus, polyomavirus, and a unique member of a new group of fish papillomaviruses in lymphocystis disease-affected gilthead sea bream. J Virol 90:8768–8779
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Perretta A, Doszpoly A, Puentes R, Bessonart M (2020) Diagnosis of lymphocystis disease in a novel host, the whitemouth croaker Micropogonias furnieri, associated with a putatively novel Lymphocystivirus species (LCDV-WC). Dis Aquat Organ 137:185–193
Tidona CA, Darai G (1997) The complete DNA sequence of lymphocystis disease virus. Virology 230:207–216
Zhang QY, Xiao F, Xie J, Li ZQ, Gui JF (2004) Complete genome sequence of lymphocystis disease virus isolated from China. J Virol 78:6982–6994
Acknowledgement
Open access funding provided by Centre for Agricultural Research.
Funding
This study was funded by the Hungarian Scientific Research Fund (OTKA K127916), by the OMA Foundation (Grant 101öu6), and by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Additional information
Handling Editor: Tim Skern.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2020_4570_MOESM1_ESM.docx
Supplementary Table 1 Name, GenBank accession number, and locus tag of the 26 core genes in the genomes of lymphocystiviruses (DOCX 16 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Doszpoly, A., Kaján, G.L., Puentes, R. et al. Complete genome sequence and analysis of a novel lymphocystivirus detected in whitemouth croaker (Micropogonias furnieri): lymphocystis disease virus 4. Arch Virol 165, 1215–1218 (2020). https://doi.org/10.1007/s00705-020-04570-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04570-1