
Abstract
Aichivirus B has been reported worldwide in calves and adult cattle with and without diarrhea. The aim of this study was to describe the molecular characteristics of the RdRP and VP1 genes of aichivirus B strains identified as the most frequent etiologic agent in a neonatal diarrhea outbreak in a high-production Brazilian dairy cattle herd. Preliminary laboratory analysis ruled out important enteropathogens (Cryptosporidium spp; Eimeria spp., E. coli F5, and bovine coronavirus). Fecal samples from diarrheic (n = 24) and asymptomatic (n = 5) calves up to 30 days old were collected for virological analysis. RT-PCR assays were performed for the detection of aichivirus B RdRP and VP1 genes and for rotavirus A VP7 and VP4 genes in fecal samples. Asymptomatic calves (control group) were negative for both viruses. Aichivirus B and rotavirus A G10P[11] genotypes were found in 54.2% (13/24) and 25% (6/24) of the diarrheic fecal samples, respectively. Aichivirus B was only identified (83.3%, 10/12) in calves up to two weeks old. Phylogenetic analysis based on the RdRP gene grouped the Brazilian strains in a new branch within the aichivirus B group. Comparative analysis of the nucleotide sequence of the VP1 gene of Brazilian and Chinese aichivirus B strains allowed the strains identified in this study to be classified in the putative lineage 1. This is the first description of a high rate of aichivirus B detection in a diarrhea outbreak in dairy calves, and the first phylogenetic study of the VP1 gene of aichivirus B wild-type strains performed in South America.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In dairy cattle herds, neonatal diarrhea is considered one of the main infectious diseases in suckling calves worldwide [1, 4]. Some classes of microorganisms, such as bacteria, protozoa, and viruses, are infectious agents and are considered determinants of neonatal diarrhea [9]. Single and mixed infections involving different classes and types of microorganisms are commonly reported [6].
Aichivirus B belongs to the family Picornaviridae, genus Kobuvirus [18]. The virions are non-enveloped with icosahedral symmetry and have a diameter of 27-30 nm and a single-stranded positive-sense RNA genome [25]. Currently, the genus Kobuvirus is divided into six species, Aichivirus A to F, the members of which can infect both humans and various species of animals [18].
The viral genome contains a leader (L) protein and has structural (VP0, VP3, and VP1) and non-structural (2A-2C and 3A-3D) regions [22]. The sequence of the 3D region of the kobuvirus genome is highly conserved and encodes the enzyme RNA-dependent RNA polymerase (RdRP). By contrast, the VP1 protein is the most exposed region of the picornavirus capsid and has a high probability of structural variation. This characteristic makes the VP1 gene the most useful one for distinguishing different members of the genus Kobuvirus [22, 25].
Aichivirus B was first identified in serum and fecal samples from healthy cattle from Japan in 2003 (U-I strain) [25]. However, since that study, aichivirus B infection has been reported in cattle herds worldwide and occurs more frequently in young animals with clinical signs of diarrhea [5, 19].
In Brazil, aichivirus B was identified in young and adult cattle in all geographical regions of Brazil in both dairy and beef herds [7, 23]. However, most reports of the occurrence of aichivirus B worldwide have been based on retrospective and transversal epidemiological surveys [7, 19, 23].
A study performed in China analyzing the nucleotide sequences of the VP1 genes of wild strains of aichivirus B showed molecular diversity, with four putative lineages (1 to 4) [8]. However, there have been no other studies based on the genetic characterization of aichivirus B strains using VP1 gene analysis.
The aim of this study was to describe the molecular characteristics of the RdRP and, particularly, the VP1 genes of wild-type aichivirus B strains that were identified as the most frequent etiologic agent in a neonatal diarrhea outbreak in calves from a high-production Brazilian dairy herd.
Materials and methods
Study location and clinical history
In early March 2015, calves with clinical signs of diarrhea from a dairy cattle herd located in the central region of the state of Paraná, southern Brazil, were evaluated. This geographical region of Paraná state is the main milk-producing region of Brazil and is notable because of the high production, technology, health, and quality levels throughout the production chain. The farm had cows of the Holstein breed managed in a free-stall system for milk production. At the time of sample collection, the farm had 930 lactating cows with two daily milkings, with an average production of 40 liters/animal/day. The nutritional and health management of the animals, including adults (cows) and young animals (heifers and calves), were adequate for the level of dairy production on the farm.
Only female calves were raised on the dairy farm, and to ensure good passive immunity, colostrum management was routinely used. For the control and prevention of neonatal diarrhea, the cows were routinely vaccinated at the end of pregnancy with inactivated commercial vaccine containing bovine rotavirus A genotype G6P[5] (UK strain), bovine coronavirus (Mebus strain), and Escherichia coli (E. coli) F5 (K99), according to the manufacturer’s instructions. However, in the summer of 2015, an outbreak of watery diarrhea, primarily in calves in the first month of life, was reported that was unresponsive to broad-spectrum antibiotic therapy. Over a period of approximately 25 days and at the time of the outbreak there were approximately 80 calves in the affected lot, with morbidity rates reaching approximately 35%.
To perform a preliminary diagnosis, five fecal samples of diarrheic calves up to 30 days of age were tested for Cryptosporidium spp. and Eimeria spp. oocysts using a modified Ziehl-Nelseen [17] and Gordon and Whitlock [15] technique, respectively. Additionally, five diarrheic fecal samples from calves up to 7 days old were analyzed by PCR assay [12] to determine the presence of enterotoxigenic E. coli F5 (K99). All diarrheic fecal samples that were initially evaluated were negative for the enteric pathogens (protozoa and bacteria) tested.
Because the animals with diarrhea were not responsive to broad-spectrum antibiotic therapy, were negative in parasitological examinations for the two main protozoa, and were negative in PCR assays for enterotoxigenic E. coli F5 (K99), the etiology of this neonatal diarrhea outbreak was determined by investigating several RNA viruses that have been recognized as enteropathogenic for calves.
Fecal samples
For virological evaluation, 24 diarrheic fecal samples from calves up to 30 days of age were collected in gloves directly from the rectum. All calves included in the study were randomly selected and had yellowish diarrhea with clinic signs of anorexia and apathy. Additionally, as a control group, five fecal samples from asymptomatic calves were collected.
Nucleic acid extraction
Fecal suspensions were prepared at 10-20% (w/v) in 0.01 M phosphate-buffered saline (PBS), pH 7.2 (137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, and 14 mM KH2PO4), and centrifuged at 3,000 × g for 5 min. RNA extraction was performed with 400 µL of fecal suspensions using a combination of the phenol/chloroform/isoamyl alcohol (25:24:1) and silica/guanidinium isothiocyanate methods [2]. The nucleic acid was eluted in 50 µL of ultra-pure RNase-free diethylpyrocarbonate-treated sterile water. Ultrapure sterile water was included as a negative control in all viral nucleic acid extraction procedures, and known positive fecal samples for each viral agent analyzed were included as a positive control [4, 21, 23].
Virological analysis
The presence of RNA for the two most frequent enteric viruses found in calf diarrheic fecal samples was investigated using molecular techniques (SN-PCR and RT-PCR) to amplify a 251-bp portion of the bovine coronavirus (BCoV) N gene [24] and 876 bp and 1,062 bp of the bovine rotavirus A (RVA) VP4 (VP8*) and VP7 gene, respectively [14, 16]. Evaluation of the presence aichivirus B RNA was performed by RT-PCR to amplify a 631- and 1,159-bp region of the RdRP and VP1 gene, respectively [8, 25]. Nuclease-free water was used as a negative control in all PCR assays. The amplified products were analyzed by electrophoresis on a 2% agarose gel in Tris-borate-EDTA buffer, pH 8.4 (89 mM Tris, 89 mM boric acid, and 2 mM EDTA), stained with ethidium bromide (0.5 µg/mL), and visualized under UV light.
Sequence analysis
RT-PCR amplification products of good quality were selected to evaluate the specificity of the test and for phylogenetic analysis. The amplicons were purified using an illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Little Chalfont, UK), quantified in a Qubit® fluorometer (Invitrogen Life Technologies, Eugene, OR, USA), and sequenced in an ABI3500 Genetic Analyzer sequencer with a Big Dye® Terminator v3.1 Cycle Sequencing Kit, using the forward and reverse primers used in the RT-PCR assay (Applied Biosystems, Foster City, CA, USA). Sequence quality analysis was performed using PHRED software, and consensus sequences were assembled using CAP3 software (http://asparagin.cenargen.embrapa.br/phph/). Sequence similarity searches were performed using the basic local alignment search tool (BLAST) program (http://blast.ncbi.nlm.nih.gov/) to determine the nucleotide sequence similarity with sequences deposited in public databases. The phylogenetic tree and the nt identity matrix were constructed using MEGA version 6.0 and BioEdit version 7.2.5 software, respectively. The analyses were based on the neighbor-joining method and the Kimura 2-parameter model. Bootstrapping was statistically supported with 1,000 replicates. The G and P genotypes of RVA were determined using the RotaC2.0 genotyping tool [20]. The referenced sequences included in this study were acquired from GenBank.
Results
All fecal samples with normal consistency (n = 5) included in this study were negative for the three evaluated viral agents. BCoV RNA was also not identified in any of the 24 diarrheic fecal samples evaluated, which allowed us to rule this important enteric virus in the etiology of the diarrhea outbreak studied.
RVA RNA was amplified in 25% (6/24) of the fecal samples from diarrheic calves; three calves had single infections, and the other three had mixed infection with aichivirus B. Sequence analysis of the G and P gene-amplification products of RVA strains identified in the diarrhea outbreak showed that the infecting RVA strains belong to the genotype G10P[11].
Of the 24 diarrheic fecal samples analyzed, 13 (54.2%) were positive for aichivirus B, but this was only identified in calves that were one or two weeks old. In this age group (8 to 14 days old), the rate of aichivirus-B-positive calves reached 83.3% (10/12). Table 1 shows the distribution of results according to the age group of calves.
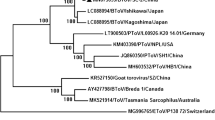
For aichivirus B, amplicon sequencing confirmed the specificity of the amplified products. Phylogenetic analysis of the RdRP genes of three aichivirus B strains (GenBank accession numbers KX587946, KX587947, and KX587948) revealed 90.7 to 90.9%, 91.6 to 92.1%, and 91.8% nt sequence identity to the aichivirus B strains described in Korea, China, and Japan, respectively (Fig. 1A).

Phylogenetic analysis of a partial nucleotide sequence (551 bp) of the RdRP gene (A) and of the complete VP1 gene (B) of Brazilian aichivirus B strains. The tree was generated using the neighbor-joining method and the Kimura 2-parameter model as the nucleotide substitution model. Bootstrap values (1,000 replicates) greater than 70% are shown. The aichivirus B strains identified in this study are indicated by a filled circle
Sequence analysis of two VP1 gene RT-PCR products allowed a comparative analysis of the sequences identified in this study with Chinese aichivirus B strains that were classified in the putative lineages proposed by Chang et al. [8]. The two Brazilian aichivirus B strains (GenBank accession numbers KX592587 and KX592588) grouped in the same branch as the prototype strain (U-I) classified as lineage 1 showed 89.2 to 89.5%, 84.6 to 86.1%, 82.7 to 83.2%, and 84.6 to 84.8% nt sequence identity and 95.1 to 95.8%, 92.5 to 93.2%, 90.6 to 91.7%, 92.5 to 93.2% amino acid (aa) sequence identity to representative aichivirus B strains from the putative lineages 1 (U-I strain), 2, 3, and 4, respectively (Fig. 1B). The two sequences described in this study showed an nt distance of 0.12 and an aa distance of 0.04 to 0.05 from the prototype strain (U-I).
Discussion
The diarrhea outbreak described in this study included diarrheic dairy calves that were irresponsive to treatment with broad-spectrum antibiotic therapy. Preliminary laboratory analyses ruled out some important enteropathogens (Cryptosporidium spp., Eimeria spp., and E. coli F5). With this information, we evaluated the probability that three known enteric RNA viruses (RVA, BCoV, and aichivirus B) were involved in the etiology of this severe diarrhea outbreak.
Although BCoV is considered an important etiological agent of neonatal diarrhea worldwide [4, 10, 13], it had no influence on this outbreak in dairy calves, because all diarrheic fecal samples included in the analysis were negative for this virus.
Throughout the world, RVA is reported to be the main viral cause of neonatal diarrhea in calves of dairy and beef cattle herds [1, 21]. In this outbreak, 25% (6/24) of diarrheic calves evaluated were RVA positive in both single (n = 3, 12.5%) and mixed (n = 3, 12.5%) infections with aichivirus B. Reports of mixed infection involving aichivirus B and other viruses are frequent, mainly with the presence of RVA and BCoV [19, 23]. Mixed infection with RVA in addition to a high frequency of aichivirus B may contribute to clinical signs of diarrhea. In general, enteric infections caused by more than one microorganism are more severe than a single infection [4], as observed in this study.
Notably, all late-pregnancy cows from this dairy farm received an inactivated commercial vaccine containing enteropathogens such as the RVA UK strain genotype G6P[5], and good colostrum management was adopted on the farm to provide adequate levels of passive immunity. However, the RVA strain that was involved in this diarrhea outbreak was classified as genotype G10P[11]. Failures in heterologous passive protection have been reported to be a cause of diarrhea by wild-type RVA strains with distinct genotypes from RVA vaccine strains in calves of both dairy and beef cattle herds [3, 21].
Aichivirus B was the main viral agent found in 54.2% (13/24) of the diarrheic fecal samples evaluated. In other studies, high positivity rates have been reported for this enteric virus, reaching 34.9% (58/166) in China [8] and 77.8% (7/9) in the Netherlands [5]. Furthermore, retrospective transversal studies conducted in Brazil revealed the presence of this enteric viral agent in 18.2% (40/222) [23] and 14.4% (31/216) [7] of the fecal samples analyzed, with a larger number of positive animals on dairy cattle farms.
Only calves up to two weeks of age were aichivirus B positive (81.25%, 13/16). Higher frequencies of detection of aichivirus B in feces of calves up to one month of age have also been reported in the Netherlands [5] and Italy [11]. The increased susceptibility to symptomatic infections in calves may be due to the immaturity of the immune system in neonatal animals compared to adults, making them more susceptible to endemic enteric microorganisms.
Phylogenetic analysis of Brazilian aichivirus B strains based on the 3D conserved genomic region encoding the viral polymerase (RdRP) reveals a difference in the position of the branches on the phylogenetic tree compared to the Japanese, Chinese, and Korean strains (Fig. 1A). However, the genomic region used for a classification of picornaviruses by molecular analysis is the VP1 capsid protein gene [22]. Therefore, the Brazilian aichivirus B VP1 gene sequences were classified in putative lineage 1, revealing greater (89.2 to 89.5%) nucleotide sequence identity to the aichivirus B prototype strain (U-I, accession numbers AB084788, NC004421) according to the classification proposed in 2014 by Chang and colleagues suggesting new genotype or lineage with a cutoff of 85.1% nucleotide sequence identity based on VP1 protein of aichivirus B. This result confirms the molecular diversity of aichivirus B strains circulating worldwide and demonstrates that the strains identified in South America exhibit molecular divergence from Chinese strains.
In conclusion, most surveys on the frequency of aichivirus B infection in cattle herds are transversal studies that include the analysis of several fecal samples from calves with or without diarrhea from different dairy and/or beef herds. To the authors’ knowledge, this is the first description of a high rate of aichivirus B identification in fecal samples from a diarrhea outbreak in dairy calves, and it is the first phylogenetic study based on the VP1 gene of aichivirus B wild-type strains performed in South America.
References
Abuelo A (2016) Investigation of an outbreak of neonatal calf diarrhoea in a dairy herd. Vet Rec Case Rep. doi:10.1136/vetreccr-2016-000372
Alfieri AA, Parazzi ME, Takiuchi E, Medici KC, Alfieri AF (2006) Frequency of group A rotavirus in diarrhoeic calves in Brazilian cattle herds, 1998–2002. Trop Anim Health Prod 38:521–526
Barreiros MA, Alfieri AF, Medici KC, Leite JP, Alfieri AA (2004) G and P genotypes of group A rotavirus from diarrhoeic calves born to cows vaccinated against the NCDV (P[1], G6) rotavirus strain. J Vet Med 51:104–109
Barry AF, Alfieri AF, Stipp DT, Alfieri AA (2009) Bovine coronavirus detection in a collection of diarrheic stool samples positive for group a bovine rotavirus. Braz Arch Biol Technol 52:45–49
Barry AF, Ribeiro J, Alfieri AF, van der Poel WH, Alfieri AA (2011) First detection of kobuvirus in farm animals in Brazil and the Netherlands. Infect Genet Evol 11:1811–1814
Brasil (2017) Ministério da Agricultura Pecuária e Abastecimento, MAPA. http://www.agricultura.gov.br/assuntos/sanidade-animal-e-vegetal/saude-animal/programas-de-saude-animal/programas-sanitarios. Acessed 18 Aug 2017
Candido M, Batinga MC, Alencar AL, de Almeida-Queiroz SR, da Gloria Buzinaro M, Livonesi MC, Fernandes AM, de Sousa RL (2017) Molecular characterization and genetic diversity of bovine Kobuvirus, Brazil. Virus Genes 53:105–110
Chang J, Wang Q, Wang F, Jiang Z, Liu Y, Yu L (2014) Prevalence and genetic diversity of bovine kobuvirus in China. Arch Virol 159:1505–1510
Coura FM, Freitas MD, Ribeiro J, de Leme RA, de Souza C, Alfieri AA, Facury Filho EJ, de Carvalho AU, Silva MX, Lage AP, Heinemann MB (2015) Longitudinal study of Salmonella spp., diarrheagenic Escherichia coli, Rotavirus, and Coronavirus isolated from healthy and diarrheic calves in a Brazilian dairy herd. Trop Anim Health Prod 47:3–11
Decaro N, Elia G, Campolo M, Desario C, Mari V, Radogna A, Colaianni ML, Cirone F, Tempesta M, Buonavoglia C (2008) Detection of bovine coronavirus using a TaqMan-based real-time RT-PCR assay. J Virol Methods 151:167–171
Di Martino B, Di Profio F, Di Felice E, Ceci C, Pistilli MG, Marsilio F (2012) Molecular detection of bovine kobuviruses in Italy. Arch Virol 157:2393–2396
Franck SM, Bosworth BT, Moon HW (1998) Multiplex PCR for enterotoxigenic, attaching and effacing, and Shiga toxin-producing Escherichia coli strains from calves. J Clin Microbiol 36:1795–1797
Fulton RW, Step DL, Wahrmund J, Burge LJ, Payton ME, Cook BJ, Burken D, Richards CJ, Confer AW (2011) Bovine coronavirus (BCV) infections in transported commingled beef cattle and sole-source ranch calves. Can J Vet Res 75:191–199
Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, Flores J, Das BK, Bhan MK (1992) Identification of group A rotavirus gene 4 types by polymerase chain reaction. J Clin Microbiol 30:1365–1373
Gordon HM, Whitlock HV (1939) A new technique for counting nematode eggs in shep faeces. In: Gonçalves PC (ed) Ueno H. Manual para Helmintoses de Ruminantes Japan International Cooperation Agency, Tokyo, pp 50–52
Gouvea V, Glass RI, Woods P, Taniguchi K, Clark HF, Forrester B, Fang ZY (1990) Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J Clin Microbiol 28:276–282
Henriksen SA, Pohlenz JFL (1981) Staining of cryptosporidium by a modified Ziehl–Neelsen technique. Acta Vet Scan 22:594–596
ICTV (2017) International Committee on Taxonomy of Viruses. https://talk.ictvonline.org/taxonomy/. Accessed 19 Aug 2017
Jeoung HY, Lim JA, Jeong W, Oem JK, An DJ (2011) Three clusters of bovine kobuvirus isolated in Korea, 2008–2010. Virus Genes 42:402–406
Maes P, Matthijnssens J, Rahman M, Van Ranst M (2009) RotaC: a web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol 9:238
Medeiros TNS, Lorenzetti E, Alfieri AF, Alfieri AA (2015) Phylogenetic analysis of a G6P[5] bovine rotavirus strain isolated in a neonatal diarrhea outbreak in a beef cattle herd vaccinated with G6P[1] and G10P[11] genotypes. Arch Virol 160:447–451
Reuter G, Boros A, Pankovics P (2011) Kobuviruses—a comprehensive review. Rev Med Virol 21:32–41
Ribeiro J, Lorenzetti E, Alfieri AF, Alfieri AA (2014) Kobuvirus (Aichivirus B) infection in Brazilian cattle herds. Vet Res Commun 38:177–182
Takiuchi E, Stipp DT, Alfieri AF, Alfieri AA (2006) Improved detection of bovine coronavirus N gene in faeces of calves infected naturally by a semi-nested PCR assay and an internal control. J Virol Methods 131:148–154
Yamashita T, Ito M, Kabashima Y, Tsuzuki H, Fujiura A, Sakae K (2003) Isolation and characterization of a new species of kobuvirus associated with cattle. J Gen Virol 84:3069–3077
Acknowledgements
This study was supported by the following Brazilian institutes: National Council of Scientific and Technological Development (CNPq – grant number 305062/2015-8), Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES), Financing of Studies and Projects (FINEP), and the Araucaria Foundation (FAP/PR). Alfieri, A.A., Alfieri, A.F. and Lorenzetti, E. are recipients of CNPq fellowships.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics statement
This study was approved and carried out in accordance with the animal ethics guidelines of the Ethical Committee for Animal Experimentation of the Universidade Estadual de Londrina.
Rights and permissions
About this article

Cite this article
Ribeiro, J., Lorenzetti, E., Júnior, J.C.R. et al. Phylogenetic analysis of VP1 and RdRP genes of Brazilian aichivirus B strains involved in a diarrhea outbreak in dairy calves. Arch Virol 162, 3691–3696 (2017). https://doi.org/10.1007/s00705-017-3531-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3531-x