
Abstract
The complete genome of ϕ16, a temperate corynephage from Corynebacterium glutamicum ATCC 21792, was sequenced and annotated (GenBank: KY250482). The electron microscopy study of ϕ16 virion confirmed that it belongs to the family Siphoviridae. The ϕ16 genome consists of a linear double-stranded DNA molecule of 58,200 bp (G+C = 52.2%) with protruding cohesive 3’-ends of 14 nt. Four major structural proteins were separated by SDS-PAGE and identified by peptide mass fingerprinting technique. Using bioinformatics analysis, 101 putative ORFs and 5 tRNA genes were predicted. Only 27 putative gene products could be assigned to known biological functions. The ϕ16 genome was divided into functional modules. Seven putative promoters and eight putative unidirectional intrinsic terminators were predicted. One site of putative «-1» programmed ribosomal frameshifting was proposed in the phage tail assembly genome region. C. glutamicum genetic tools could be broadened by exploiting the known integrase gene (gp33) and the newly identified excisionase gene (gp47), participating in site-specific recombination between ϕ16-attP/attB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Corynebacterium glutamicum is widely used to produce commercially interesting bio-based substances [1]. Phages present a problem for the biotechnology industry and cause financial losses. Many corynephages have been isolated, but only a few of them have been completely sequenced (e.g. [2, 3]). In the present study, the genome of ϕ16, a temperate corynephage from C. glutamicum (ATCC 21792), kindly provided by Dr. Trautwetter [4], was sequenced and annotated. This information could provide valuable evolutionary insights and be helpful for phage-resistant strain construction [5]. Different integrative vectors targeting different attB-sites have been constructed based on known integrases of phages ϕAAU2 [6], beta [7], ϕ304L [8] and ϕ16 [9] from C. glutamicum strains. The newly identified ϕ16 excisionase, in addition to the known integrase gene, could be useful for broadening C. glutamicum genetic tools, e.g. for site-specific integration/excision of DNA fragments into bacterial chromosomes, as was demonstrated for other phage-based systems [10].
Phage ϕ16 was induced from the natural lysogen, C. glutamicum ATCC 21792, propagated and purified by centrifugation in CsCl-gradient as described [4, 11].
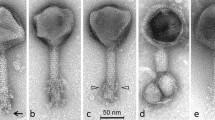
Transmission electron microscopy study of ϕ16 virion confirmed that it belongs to the family Siphoviridae, with a polyhedral head of 73 nm in width and 336 nm in length, and with a non-contractile striated tail of 14 nm in diameter (Fig. 1a), in line with Dr. Trautwetter’s group data [4]. Subsequently, one of the putative ϕ16 gene products (gp), gp16, was assigned to the tail tape measure protein (TMP). The relationship between observed tail length (~336 nm) and TMP size (2,151 aa), with a ratio of 0.156 nm/aa, is reasonable [12].

(a) Electron micrograph of ϕ16. Bar, 500 nm. (b) SDS-PAGE analysis of ϕ16 structural proteins. Molecular weight markers (lane I). Protein profile of ϕ16 (lane II). (c) Four major bands underwent peptide mass fingerprinting analysis; the corresponding predicted amino acid sequences (not highlighted) and the aa sequences detected in the analysis (highlighted) are shown
Purified phage DNA was hydrodynamically sheared, and fragments of 2 to 5 kb in size were blunted and then cloned and sequenced by the Sanger’s method. A total of 346 individual DNA fragments were sequenced with an average length of 750 ± 130 bp, and an achieved sequence coverage of ~ 6.6-fold. Closure of gaps was accomplished by primer walking. The genome sequence was finalized by determining the cos sequence with a sequence run-off experiment and comparison of the nucleotide sequence with the ligated phage ends.
Three bioinformatic on-line programs were used for ORFs prediction in the ϕ16 genome: Open Reading Frame Finder (https://www.ncbi.nlm.nih.gov/orffinder/), Glimmer 3 (http://www.ncbi.nlm.nih.gov/genomes/MICROBES/glimmer_3.cgi) and GeneMark S (http://exon.biology.gatech.edu/). The putative tRNAs genes were predicted using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/). Two web services, the phiSITE PromoterHunter (http://www.phisite.org/main/index.php?nav=tools&nav_sel=hunter) (with parameters for “–10” and “–35” [Supplementary Fig. 1]) and the PePPER tool-box (http://pepper.molgenrug.nl) were used to search for putative promoters. Intrinsic terminators were identified with “ARNold finding terminators” (http://rna.igmors.u-psud.fr/toolbox/arnold/index.php) with additional evaluation of free energy parameters by the online version of the ViennaRNA package (http://rna.tbi.univie.ac.at/).
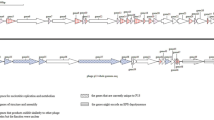
The ϕ16 genome is a double-stranded DNA molecule of 58,200 bp in length (G + C = 52.2%) with 3′-protruding single-stranded cohesive ends of 14 nt (3′-GGAAGGTGGAGGCT and CCTTCCACCTCCGA-3′). Using bioinformatics analysis, 101 putative ORFs covering ~92.4% of the total DNA length were identified. Only 27 gp(s) could be assigned to known biological functions (Supplementary Table 1); the other 55 gp(s) displayed homology to hypothetical proteins, and 19 gp(s) had no homologues in the databases (Fig. 2). Seven putative promoters (1 – leftward and 6 – rightward) and eight putative intrinsic unidirectional terminators (1 – leftward and 7 – rightward) were predicted in the intergenic spaces of the ϕ16 genome (Fig. 2, Supplementary Table 2, 3). Five tRNA Lys(UUU), Arg(UCU), Asn(GUU), Tyr(GUA) and Trp(CCA) were identified (Supplementary Fig. 2). Comparison between ϕ16 and C. glutamicum codons frequency support the hypothesis that phage-encoded tRNAs could compensate codon frequency bias and promote efficient translation of phage-derived mRNA [13] (Supplementary Fig. 3).

Genomic organization of ϕ16 phage. ORFs are numbered consecutively from left to right and are indicated by arrows (or triangles) in the direction of transcription. ORFs, joined by braces, are provided for the proposed functional modules of the ϕ16 genome. Promoter positions and directions are indicated by thin arrows; intrinsic terminators and tRNAs are depicted as dark and light boxes, respectively
Based on homology to known phage proteins, functional domains, and mutual arrangement, putative functions were assigned to products of 27 predicted ORFs (Supplementary Table 1). The entire genome was divided into the six functional modules (Fig. 2). The DNA packaging module contains small and large terminase subunits (gp2 and gp3) and the portal protein (gp4). The prohead protease (gp5), the major capsid and tail proteins (gp7 and gp13), the tail TMP (gp16), and the tail fiber protein (gp20) could be predicted in the structural components and assembly module. At the same time, four major structural proteins, the major capsid and tail proteins (gp7 and gp13) and two proteins with unknown function (gp6 and gp12), were detected by SDS-PAGE and identified by trypsin-based peptide mass fingerprinting technique (PMF), using Ultraflex II LC-MALDI-TOF/TOF (Brucker), performed according to Govorun et al. [14]. Furthermore, detection of N-terminal Met residue retention in a trypsin-digested peptide from gp12 and its elimination from N-terminal peptides from gp6 and gp7 confirmed the N-terminal processing rule [15] (Fig. 1b, c).
A putative site of a –1 programmed ribosomal frameshifting (PRF) could be found in the proposed tail assembly genes and was composed of three functional elements: an internal SD (5’–GAGG→3’), a “slippery sequence” (5′–GGGGGAA→3′) and an H-type pseudoknot RNA structure (Supplementary Fig. 4) [16]. The PRF was predicted to lead to the formation of a large fusion protein, gp14A.
Homologues of two known enzymes were predicted in the host lysis module: the endolysin (gp22) and the holin (gp23). The lysogeny control module was unusual: it contained two putative integrases (gp33 and gp28), the excisionase (gp47), the phage superinfection exclusion protein (gp34), and the transcriptional regulator (gp36). The nucleotide sequence of the ϕ16 int gene (corresponding to ORF33 in our annotation) and the ϕ16 attP site were deposited previously (GenBank: Y12471.1) [8] and differ from the newly sequenced ORF33 in several points due to sequencing errors in the past, that resulted in differences in the structures of the corresponding gp(s) (Supplementary Fig. 5). We confirmed the ability of gp33 to provide site-specific integration of recombinant DNA into the ϕ16-attB of the C. glutamicum ATCC 21792c chromosome, which was previously shown by the Trautwetter group [9]. We also demonstrated experimentally the effective excision of integrated recombinant DNA when gp33 and gp47 are expressed simultaneously of (manuscript in preparation). The experiments also showed that the second putative integrase, gp28, could not use the previously established ϕ16-attP site [8] for site-specific recombination. No other putative attP-site was detected in the vicinity of ORF28 (unpublished result).
The replication/recombination/metabolism module also contained homologues to known proteins: ParB-like protein (gp42), HNH homing endonuclease (gp52), the transcriptional regulator (gp56), SSB protein (gp58), glutaredoxin (gp62), RusA endodeoxyribonuclease (gp63), methyltransferase (gp64), chromosomal partitioning protein (gp68), oligoribonuclease (gp94) and ATPase (gp101).
The analysis indicated that some modules of the ϕ16 genome had complete or partial homology to distinct chromosomal regions of four bacteria, leading us to hyphothesize that these are uncharacterized prophages in bacterial genomes. Throughout large parts of the genome sequence, significant similarity was observed between ϕ16 and the hypothetical prophage Corynebacterium pyruviciproducens ATCC BAA-1742, at the nucleotide and deduced protein sequence level. Significant similarity was also observed, throughout the whole genome, between ϕ16 and the hypothetical prophage Brevibacterium flavum ATCC 15168 (identical to prophage of C. glutamicum ATCC 14067). We also observed similarity between predicted ϕ16 proteins, which are involved in DNA packaging, head and tail structural components, as well as assembly modules, and proteins of a Corynebacterium ulcerans BR-AD22 hypothetical prophage. Several ϕ16 lysogeny control genes (ORFs 30, 33, 35, 47) were very similar, at the protein and nucleotide level, to genes of the hypothetical prophage of C. falsenii DSM 44353, strain BL 8171. Furthermore, a significant part of the ϕ16 genome, as well as the second part of the lysogeny control genes (ORFs 28, 29, 34, 36) share homology with a hypothetical prophage from C. pyruviciproducens ATCC BAA-1742. To demonstrate the general sequence homology with the four hypothetical prophages, multi-dot plots were obtained (Supplementary Fig. 6). Neither protein nor nucleotide homology was observed between ϕ16 and BFK20 or P1201. However, the ϕ16 genome is not present in any bacterial chromosome in the database; therefore, the sequence of the entire ϕ16 phage genome was deposited for the first time in GenBank, under accession number KY250482.
References
Becker J, Wittmann C (2012) Bio-based production of chemicals, materials and fuels—Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. doi:10.1016/j.copbio.2011.11.012
Bukovska G, Klucar L, Vlcek C, Adamovic J, Turna J, Timko J (2006) Complete nucleotide sequence and genome analysis of bacteriophage BFK20—a lytic phage of the industrial producer Brevibacterium flavum. Virology 348:57–71. doi:10.1016/j.virol.2005.12.010
Chen CL, Pan TY, Kan SC et al (2008) Genome sequence of the lytic bacteriophage P1201 from Corynebacterium glutamicum NCHU 87078: Evolutionary relationships to phages from Corynebacterineae. Virology 378:226–232. doi:10.1016/j.virol.2008.05.027
Moreau S, Leret V, Marrec CL, Varangot H, Ayache M, Bonnassie S, Blanco C, Trautwetter A (1995) Prophage distribution in coryneform bacteria. Res Microbiol 146:493–505. doi:10.1016/0923-2508(96)80295-6
Labrie SJ, Samson JE, Moineau S (2010) Bacteriophage resistance mechanisms. Nat Rev Microbiol 8(5):317–327. doi:10.1038/nrmicro2315
LeMarrec C, Moreau S, Loury S, Blanco C, Trautwetter A (1996) Genetic characterization of site-specific integration functions of ϕAAU2 infecting “Arthrobacter aureus” C70. J Bacteriol 178:1996–2004
Oram M, Woolston JE, Jacobson AD, Holmes RK, Oram D (2007) Bacteriophage-based vectors for site-specific insertion of DNA in the chromosome of Corynebacteria. Gene 391(1–2):53–62. doi:10.1016/j.gene.2006.12.003
Moreau S, Le Marrec C, Blanco C, Trautwetter A (1999) Analysis of the integration functions of & #x03D5;304L: an integrase module among corynephages. Virology 255:150–159. doi:10.1006/viro.1998.9524
Moreau S, Blanco C, Trautwetter A (1999) Site-specific integration of corynephage ϕ16: construction of an integration vector. Microbiology 145:539–548. doi:10.1099/13500872-145-3-539
Haldimann A, Wanner BL (2001) Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi:10.1128/JB.183.21.6384-6393.2001
Boulanger P (2009) Purification of bacteriphages and SDS-PAGE analysis of phage structural proteins from host particles. Methods Mol Biol 502:227–238. doi:10.1007/978-1-60327-565-1_13
Abuladze NK, Gingery M, Tsai J, Eiserling FA (1994) Tail length determination in bacteriophage T4. Virology 199(2):301–310. doi:10.1006/viro.1994.1128
Bailly-Bechet M, Vergassola M, Rocha E (2007) Causes for the intriguing presence of tRNAs in phages. Genome Res 17:1486–1495. doi:10.1101/gr.6649807
Govorun VM, Moshkovskii SA, Tikhonova OV et al (2003) Comparative analysis of proteome maps of Helicobacter pylori clinical isolates. Biochemistry (Mosc) 68:42–49
Eggeling L, Bott M (2005) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton
Xu J, Hendrix RW, Duda RL (2004) Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell 16(1):11–21. doi:10.1016/j.molcel.2004.09.006
Acknowledgements
The authors are very grateful to Drs. Andrey O. Lobanov, Sergey V. Smirnov, and Yurges A.V. Yomantas for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lobanova, J.S., Gak, E.R., Andreeva, I.G. et al. Complete nucleotide sequence and annotation of the temperate corynephage ϕ16 genome. Arch Virol 162, 2489–2492 (2017). https://doi.org/10.1007/s00705-017-3383-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3383-4