
Abstract
Porcine epidemic diarrhea virus (PEDV) first emerged in Vietnam in 2009. In this study, the complete genomes of three Vietnamese PEDV isolates were characterized. These three isolates were isolated from 3-day-old pigs experiencing diarrhea. Two isolates were from swine farms in the south, and the other was from northern Vietnam. The whole genome sequences of these isolates are 28,035 nucleotides in length and have characteristics similar to those of other PEDV isolates. All three Vietnamese PEDV isolates share 99.8 % and 99.6 % sequence identity at the nucleotide and amino acid level, respectively, and have insertions of four amino acids (GENQ) and one amino acid (N) at positions 56-59 and 140, respectively, and one deletion of two amino acids (DG) at positions 160-161. Phylogenetic analysis based on the whole genome revealed that the three Vietnamese PEDV isolates are grouped together with new variants from China from 2011 to 2012 and are genetically distinct from US isolates and the classical PEDV variant. The results suggest that Vietnamese PEDV isolates are new variants, as evidenced by their genetic composition of insertions and a deletion in the spike gene, and they might have originated from the same ancestor as the Chinese PEDV strain. This study provides a better understanding of the molecular characteristics of PEDV in Vietnam.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine epidemic diarrhea (PED) is a devastating enteric disease characterized by vomiting, acute watery diarrhea, and severe dehydration leading to death [1, 2]. Although pigs of all ages are susceptible and display high mobility, the disease causes particularly high mortality in piglets less than one week of age [3]. Mortality in older pigs, however, is lower. PED was first recognized in 1976 [2, 4]. Since its first emergence, PED has continued to have a severe economic impact in many countries worldwide, and it poses a threat to countries where PED has not been reported.
PED virus (PEDV) is the causative agent of PED [1]. PEDV is an enveloped, positive-sense, single-stranded RNA virus belonging to the genus Alphacoronavirus, family Coronaviridae, order Nidovirales [2]. The PEDV genome is approximately 28 kb in length and is composed of seven open reading frames (ORFs) [5]. ORF1a and ORF1b cover 70 % of the entire genome, encoding the non-structural replicase gene. Five coding regions downstream of the replicase gene encode four structural proteins, including the spike (S), envelope (E), membrane (M) and nucleocapsid (N) proteins. ORF3 encodes an accessory protein and is located between the genes for the structural proteins. The S and ORF3 proteins play important roles in the pathogenesis of the disease [6]. The S protein, a glycosylated protein, extends from the viral surface [7]. This protein is involved in the pathogenesis of disease by binding to cell receptors on the host cell membrane and penetrating cells via membrane fusion [8, 9]. The S protein contains epitopes capable of inducing neutralizing antibodies [10–13] and has been reported to have a high degree of genetic diversity [14–17]. The ORF3 gene encodes accessory proteins whose functions are less well known. The role of the ORF3 gene in the virulence of PEDV has received much attention, because virulence can be reduced by altering this gene through cell culture adaptation [18, 19]. The ORF3 gene has been used in several reports to differentiate between field- and vaccine-derived isolates [19]. The other two genes encode the M and E proteins, the former of which is associated with the virus-assembly process [6, 20, 21]. The N protein, which binds to virion RNA and provides a structural base for the helical nucleocapsid, is a basic phosphoprotein associated with the genome [22–24].
PED was first recognized in Belgium and the United Kingdom from 1976 to 1978 [2]. Since the emergence of PED, disease outbreaks have been reported in several European countries, including Switzerland, Germany, France, the Netherlands, and Bulgaria [2, 4]. In Asia, PED was first identified in Japan in 1982 [25], China in 1986 [26], India in 2003, and Thailand in 2007 [27]. Unlike in Europe, PED is considered an endemic pathogen in Southeast Asia and has continued to cause a devastating enteric disease and severe economic losses since its first emergence. However, the emergence of PED in North America in 2013 has established PEDV as one of the most serious enteropathogenic diseases threatening the swine industry worldwide.
PED first emerged in Vietnam in 2009 [28]. The disease was first observed in the southern provinces of Vietnam without any known route of introduction. Soon after emergence, the disease spread throughout the major swine-producing regions in Vietnam, including the northern, central and the southern regions. At present, PED has developed to an endemic stage, causing sporadic outbreaks. Vietnamese PEDV isolates from farms in the southern provinces from 2009 to 2010 have been reported to be closely related to Chinese isolates [28]. However, this previous study investigated only partial S and M gene sequences. Therefore, complete genome sequencing data of PEDV in Vietnam are needed to understand genetic diversity, establish a reference genome for PEDV isolates in Vietnam, and identify the source of introduction of the virus. In this study, whole-genome sequences are reported for three PEDV strains isolated from pigs displaying severe diarrhea from farms located in the northern and southern provinces of Vietnam. These results provide an improved understanding of the molecular characteristics of PEDV in Vietnam and aid in the design of successful prevention and control programs, including the selection of isolates used for vaccine development in the near future.
Materials and methods
Viral isolation
Intestinal samples from 3- to 4-day-old pigs displaying clinical features associated with PED, including vomiting, watery diarrhea and dehydration, were collected from three different herds that experienced PED outbreaks in 2013. One herd was located in northern Vietnam, and the other two were located in southern Vietnam. The intestinal samples were subjected to PEDV isolation using the continuous Vero cell line (ATCC, CCL-81) [29]. Briefly, the intestinal samples of piglets were minced into small pieces and suspended in 10 ml of phosphate-buffered saline (PBS; 0.1 M, pH 7.2). The suspended samples were vortexed and clarified by centrifugation at 1,350 g for 10 minutes. The supernatant was filtered through 0.45-µm filters and stored at -80 °C until use.
Five hundred microliters of supernatant samples was diluted with maintenance media (MM) consisting of Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10 µg of trypsin (Gibco, Grand Island, NY, USA) per ml and inoculated into a 25-cm2 flask that had been previously plated with Vero cells for 2 days or until full confluence was reached. The inoculated 25-cm2 flask was incubated at 37 °C in 5 % CO2 for 1 hour. Then, the medium was replaced with 10 ml of MM and incubated again for a period of 2-3 days. The cytopathic effect (CPE) was observed with a light microscope. Then, the inoculated flasks were frozen and thawed twice before being centrifuged at 1,350 g for 10 minutes to collect the supernatant, which was stored at -80 °C. The intestinal samples were considered negative for PEDV isolation following three blind passages.
Reverse transcription-polymerase chain reaction
Viral RNA was extracted from the infected Vero culture supernatant using a Nucleospin® viral RNA isolation kit (Macherey-Nagel Inc., Duren, Germany) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from the extracted RNA using M-MuLV Reverse Transcriptase (New England Biolabs Inc., Ipswich, MA, USA). Twelve pairs of the oligonucleotide primers from a previous study were used to amplify the different regions of the Vietnamese PEDV [30]. PCR amplification was performed using cDNA with GoTaq® Green Master Mix (Promega, Madison, WI, USA) according to the manufacturer’s protocol under the conditions listed in Table 1.
Cloning, plasmid purification and sequence determination
The PCR products were purified and cloned into pGEM-T® Easy Vector for the subsequent transformation of Escherichia coli using a commercial kit (Promega, Madison, WI, USA). Bacterial transformant colonies were randomly selected from each sample for plasmid purification using a Nucleospin® Plasmid kit (Macherey-Nagel, Duren, Germany) according to the manufacturer’s instructions and sequenced in both directions in triplicate using an ABI Prism 3730XL sequencer. The 5′-terminal sequences were determined by 5′ rapid amplification of cDNA ends (RACE) [31].
Sequence analysis
Nucleotide and deduced amino acid sequence alignments were performed using the CLUSTAL W program [32]. Phylogenetic analysis was performed based on the nucleotide sequences in the entire genome and the ORF1, complete S, ORF3, E, M, and N genes of three PEDV isolates in this study along with 35 other PEDV isolate sequences available in GenBank (Supplementary Material 1) using the MEGA5 program [33]. A midpoint-rooted neighbor-joining tree was generated based on the Kimura 2-parameter model. The robustness of the phylogenetic analysis and significance of the branch order were determined by bootstrap analysis with 1,000 replicates. The percentages of sequence identity between the isolates at the nucleotide and amino acid level were calculated.
Results
Virus
Three PEDV isolates were isolated from intestinal samples of 3-day-old pigs displaying severe watery diarrhea and dehydration, using Vero cells. A cytopathic effect characterized by cell fusion and syncytium formation was observed following isolation in Vero cells. Three isolates, designated VN/KCHY-310113 (accession number KJ960180), were taken from swine farms in northern Vietnam in January 2013, and two strains, designated VAP1113-1 (accession number KJ960179) and JFP1013-1 (accession number KJ960178), were taken from two swine farms in southern Vietnam in October and November 2013.
Phylogenetic analysis and sequencing data
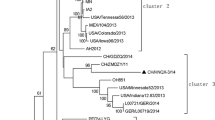
To investigate the genetic relationship among these isolates, a phylogenetic tree based on the whole genome of the nucleotide sequences of the three Vietnam PEDV isolates along with 35 PEDV isolate sequences from other countries was constructed. The phylogenetic tree demonstrated the evolution of all 35 PEDV isolates into two distinct groups, designated groups G1 and G2 (Fig. 1A). Group G1 consisted of nine isolates: the prototype CV777, three isolates from Korea (SM98 and virulent and attenuated DR13), and five isolates from China (SD-M, LZC, CH/S, attenuated vaccine, and JS2008). Group G2 consisted of isolates from Vietnam, Korea, China, and the United States. Group G2 was further divided into two subgroups, consisting of G2-1 and G2-2. Three Vietnamese PEDV isolates were included in subgroup G2-1 along with seven isolates from China from 2011-2012 (LC, AJ1102, ZJCZ4, GD-1, GD-A, CHGD-01 and CH/GDGZ/2012). Subgroup G2-2 included seven isolates from China (2011-2012), eight isolates from the United States (2013), and four isolates from Korea isolated during the period from 2013 to 2014 (KNU-1305, KUIDL-PED-2014-007, KUIDL-PED-2014-002, and KUIDL-PED-2014-001) that were reported to be PEDV strains resembling those found in the United States.

Phylogenetic analysis of porcine epidemic diarrhea virus based on nucleotide sequences of the whole genome (A), ORF1 (B), spike (C), ORF3 (D), envelope (E), membrane (F) and nucleocapsid (G) genes. Trees were constructed by the neighbor-joining method with bootstrapping for 1,000 replicates. Red dots represent the three Vietnamese isolates
To analyze the genetic diversity of the isolates, the percentage of similarity between isolates based on the whole genome was calculated at the nucleotide and amino acid level. The three PEDV isolates from Vietnam shared the highest genetic similarities with Chinese isolates in subgroup G2-1 (LC, ZJCZ4, CHGD-01, CH/GDGZ/2012, GD-1, GD-A, and AJ1102), with nucleotide and amino acid sequence identities ranging from 98.4 % to 98.9 % and 95.9 % to 97.1 %, respectively. The three PEDV isolates from Vietnam shared only 97.0 %-98.1 % and 93.9-95.3 % identity at the nucleotide and amino acid level, respectively, with subgroup G2-2. In addition, the genetic similarity between three isolates from Vietnam and isolates in Group G1 was 96.0-97.3 % and 90.2-93.3 % at the nucleotide and amino acid level, respectively.
The whole genome of the three PEDV isolates from Vietnam characterized in this study was 28,035 nucleotides (nt) in length. The nucleotide and deduced amino acid sequences of the whole genome of the three Vietnamese PEDV isolates were aligned with those of 35 PEDV isolates. The sequence alignments demonstrated that the three PEDV isolates from Vietnam have a genome organization similar to that of other PEDV isolates reported previously [5, 6], which are characterized by the gene order ORF1a/1b-S-ORF3-E-M-N. The ORF1a/1b is 20,344 nucleotides in length and is the largest ORF. Genes ORF1a (nt 297- 12602) and ORF1b (nt 12601-20637) cover two-thirds of the genome. The ORF1 of the three Vietnamese PEDV isolates has 91.4 %-97.1 % amino acid sequence identity to other isolates, exhibiting the highest identity (96.0 %-97.1 %) to Chinese strains (CH/GDGZ2012, GD-1, GD-A, ZJCZ4, AJ1102, CHGD-01 and LC). There are no insertions or deletions in the sequence of this ORF1. Compared to other PEDV strains, the amino acid sequence identities of ORF1a and ORF1b of all Vietnamese PEDV isolates are 91.2 %-96.7 % and 91.6 %-97.9 %, respectively. A phylogenetic tree based on ORF1 gene nucleotide sequences demonstrated that all three PEDV isolates along with 35 other PEDV isolates evolved into two groups, namely, G1 and G2 (Fig. 1B). Group G1 consisted of the prototype CV777, four isolates from China (SD-M, JS2008, LZC and attenuated vaccine) and two from Korea (SM98, and attenuated DR13). Three Vietnamese isolates were classified into group G2 along with 15 isolates from China, eight from the US and five from Korea, with 97.3 %-98.9 % nucleotide sequence identity and 94.1 %-97.1 %amino acid sequence identity.
The S gene, which encodes the S protein, follows ORF1 in the genome sequence. The S gene of the three PEDV isolates from Vietnam is 4,158 nucleotides in length, encoding a protein of 1,386 amino acids. A phylogenetic tree based on the complete S gene sequence demonstrated that all three PEDV isolates, along with 35 other PEDV isolates, evolved into two groups [14, 30], namely, G1 and G2 (Fig. 1C), similar to the classification of the whole genome. Group G1 consisted of the prototype CV777, five isolates from China (CH/S, SD-M, JS2008, LZC and attenuated vaccine) and three from Korea (SM98, virulent and attenuated DR13). Group G2 was further divided into three subgroups, designated G2-1, 2-2, and 2-3. As noted above, the PEDV isolates in group G2 have unique characteristics, including one deletion position and two insertion positions in the spike genes, making them genetically distinct from isolates in group G1. The three PEDV isolates from Vietnam clustered into subgroup G2-3 along with eight isolates from China that were isolated during the period from 2011 to 2012, with nucleotide and amino acid sequence identity of 97.1 %-98.6 % and 96.9 %-98.2 %, respectively. When compared to subgroup G2-2, the nucleotide and amino acid sequence identities of subgroup G2-3 are 96.7 %-98.7 % and 96.4 %-99.0 %, respectively. When compared to Group G1, the nucleotide and amino acid sequence identities of subgroup G2-3 are 92.2 %-94.6 % and 90.9 %-94.0 %, respectively. In addition, the three PEDV isolates from Vietnam clustered distinctly from the US-PEDV isolates.
The deduced amino acid sequence demonstrated that all three PEDV isolates from Vietnam have unique characteristics in the S gene, including deletion and insertion mutations in the spike gene. The S protein of three Vietnamese PEDV isolates possesses two insertions of four (56GENQ59) and one (140N) amino acids at positions 56-59 and 140, respectively, and one deletion of two amino acids (160DG161) at positions 160-161 (Supplementary Material 2). The insertion and deletion are located in the hypervariable domain in the N-terminus of the S1 region, indicating that the Vietnamese isolates are genetically similar to isolates that were isolated during 2011-2012 in China that are considered new variants.
The four subsequent genes in the sequence are the ORF3, E, M and N genes. The ORF3 of Vietnamese PEDV isolates is 675 nucleotides in length and encodes a protein of 225 amino acids. A phylogenetic analysis based on the ORF3 gene revealed that all PEDV isolates were divided into four groups, designated G1, G2, G3 and G4 (Fig. 1D). Group G1 consisted of the prototype CV777, SM98 and LZC. Group G2 contained three Chinese isolates (JS2008, SD-M and attenuated vaccine) and attenuated DR13, which is a vaccine isolate from Korea. Isolates in Group G2 have the unique characteristic of a 17-amino-acid deletion at positions 82-98. Group G3 included isolates from the United States and China as well as five Korean field isolates (Virulent-DR13, KNU-1305, KUIDL-PED-2014-007, KUIDL-PED-2014-002, KUIDL -PED-2014-001). All three PEDV isolates from Vietnam were in Group G4, along with seven Chinese isolates. Isolates in Group G4 possess four unique amino acid substitutions (L25S, I70V, C107F and D168N) compared to isolates in the other groups. The VN/KCHY-310113/2013 isolate has two more amino acid changes (F57L and C100R) and an amino acid substitution (I203V) in JFP10113-1 and VAP1113-1 compared to the other isolates. Sequence analysis showed that the three Vietnamese PEDV isolates share 98.5 %-99.4 % and 98.2 %-99.5 % nucleotide and amino acid sequence identity with other strains in group G4. When the three Vietnamese strains were compared to those of group G3, the nucleotide and amino acids sequence identities were 95.5 %-97.0 % and 95.9 %-97.3 %, respectively. When these strains were compared to those of group 2, the nucleotide and amino acid sequence identities were 89.3 %-89.7 % and 89.2 %-90.1 %, respectively. The nucleotide and amino acid sequence identities were 91.5 %-96.2 % and 89.7-95.0 %, respectively, when compared to group 1.
The E gene of PEDV isolates from Vietnam has 231 nucleotides, encoding a protein of 77 amino acids. A phylogenetic analysis based on the E protein demonstrated that all PEDV isolates were classified into two distinct groups. PEDV isolates from Vietnam were in group 2 along with the recent PEDV from China and the United States (Fig. 1E). All three PEDV isolates from Vietnam showed an amino acid change (T51I) compared to other isolates. The nucleotide and amino acid sequence identities of Vietnamese PEDV strains are 97.8 %-99.5 % and 97.3 %-98.6 % compared to other isolates in group G2. When the Vietnamese strains were compared to those of group G1, the nucleotide and amino acid sequence identities were 88.3 %-97.8 % and 85.5 %-97.3 %, respectively.
The M gene of all Vietnamese PEDV isolates is 681 nucleotides in length and encodes a protein of 226 amino acids. No insertion or deletion mutations were found in the M genes of these isolates. Phylogenetic analysis indicated that all PEDV isolates were separated into two groups (Fig. 1F). Group 1 consisted of CV777, five isolates from China (SD-M, LZC, CH/S, attenuated vaccine and JS2008) and two isolates from Korea (SM98 and attenuated DR13). Three Vietnamese PEDV isolates were in subgroup 2-1 along with seven Chinese isolates (CH/GDGZ/2012, CHGD-01, GD-A, GD-1, LC, ZJCZ4 and AJ1102), with 99.1 %-100 % amino acid sequence identity. These isolates also have an amino acid change (G192S) when compared to other groups. Group 2-2 contains all of the US isolates, six isolates from China (CH/FJZZ-9/2012, GD-B, JS-HZ2012, AH2012, BJ-2011-1 and CH/FJND-3/2011) and four isolates from Korea (KNU-1305, KUIDL-PED-2014-007, KUIDL-PED-2014-002, KUIDL-PED-2014-001). This group was characterized by two amino acid changes (E13Q and A214S). All PEDV isolates in Group 2 shared an amino acid mutation from V to A at amino acid position 42. The virulent isolate DR13 from Korea clustered separately in subgroup 2-3, as it possessed an additional amino acid change (F43L). Sequence analysis revealed that the Vietnamese PEDV strains shared 99.5 %-99.8 % and 99.1 %-100 % identity at the nucleotide and amino acid level, respectively, with Chinese strains in subgroup G2-1. When the Vietnamese PEDV strains were compared with those in Group G2-2, the nucleotide and amino acid sequence identities were 98.3 %-98.8 % and 98.2 %-98.6 %, respectively. When these strains were compared with those in group G1, the nucleotide and amino acid sequence identities were 96.6 %-98.6 % and 96.5 %-99.1 %, respectively.
Sequence analysis of the N gene revealed that all Vietnamese PEDV isolates are 1,326 nucleotides in length and encode a protein of 441 amino acids. These isolates have 95.6 %-98.7 % nucleotide and 95.0 %-97.9 % amino acid sequence identity to other isolates, and the highest homology is to CHGD-01 from China at the nucleotide (98.7 %) and amino acid (97.7 %-97.9 %) levels. A phylogenetic tree based on the nucleotide sequences of the N gene indicated that all PEDV isolates were classified into two groups, designated G1 and G2 (Fig. 1G). The three Vietnamese PEDV isolates were in subgroup G2 along with six Chinese isolates (CH/GDGZ2012, CHGD-01, GD-1, LC, AJ1102, and GD-A), with nucleotide and amino acid sequence identities ranging from 97.4 % to 98.7 % and from 95.0 % to 97.9 %.
When compared to Group G1, these strains shared 95.6 %-97.5 % and 95.9 %-97.9 % nucleotide and amino acid sequence identity, respectively. There were no insertions or deletions in this region. Compared to other isolates, all of the Vietnamese PEDV isolates except VN/KCHY-310113 possessed six amino acid substitutions (N146S, V216M, P248L, K312R, G313V, and T356I) and one amino acid change from G to P at amino acid position 84 compared to the CV777 strain.
Discussion
PED first emerged in Vietnam in 2009. Since its emergence, the disease has developed to an endemic stage, continuing to cause economic damage to the Vietnamese swine industry. A previous report suggested that PEDV isolates in Vietnam were closely related to Chinese isolates [28]. The genetic analysis in that study, however, was based on partial S and M gene sequences only. An analysis based on partial S gene sequences alone might skew the epidemiological investigation and evolution studies of the virus. A previous study from China indicated that the emergence of a new variant of PEDV was responsible for the recent outbreak in China from 2010 to 2011 [34], suggesting that there have been two variants of PEDV in China. The new variant is genetically distinct from the old variant in the spike gene regions, with two insertions of four (56GENQ59) and one (140N) amino acids at positions 56-59 and 140, respectively, and a deletion of two amino acids (160DG161) at positions 160-161. The analysis of partial S gene sequences using the primers reported would not have covered the insertion and deletion regions that would allow for differentiation between classical and new variants. Thus, the question remains as to what type of PEDV currently exists in Vietnam. In this study, we determined the complete genome sequences of three PEDV strains isolated from pigs exhibiting severe diarrhea in swine herds in the northern and southern regions of Vietnam. The results reported here provide a reference genome sequence that can provide guidance for PEDV control in Vietnam.
For the characterization of PEDV isolates, a phylogenetic tree was constructed based on the complete genome of PEDV isolated from Vietnam and 35 other isolates whose genome sequences were available in GenBank. Deduced amino acid sequences of the complete genome were also investigated. Based on phylogenetic analysis of the complete genome sequence, three PEDV isolates from Vietnam were grouped together in group G2 along with PEDV isolates from China (isolated after 2010), Korea and the United States. This cluster is genetically distinct from cluster 1, into which CV777, attenuated vaccine isolates and isolates from China (isolated before 2010) and Korea were grouped. The differences between these two clusters are based on ORF1 and the spike gene regions [5, 14, 34–36]. Cluster 2 was further divided into two sub-clusters, designated 2-1 and 2-2. Three PEDV isolates from Vietnam were grouped into sub-cluster 2-1. This sub-cluster includes, in addition to Vietnam isolates, isolates from China that were reported to be a new variant. These isolates were separated from Chinese (AH2012), US and US-like Korean isolates, which were included in sub-cluster 2-2. These findings suggest that the PEDV strains isolated in Vietnam are new variants. In addition, the results of genetic analysis suggest a close relationship between Vietnamese isolates and new variants of Chinese PEDV isolates from 2011 to 2012.
In 2014, several reports from Asian countries, including Korea, Taiwan and Japan [37, 38], suggested that there has been an emergence of US-like PEDV in Asian countries. The results of our study, however, demonstrate that at present, PEDV in Vietnam is not influenced by US-like PEDV strains. PED is endemic in Korea, Japan and Taiwan. The emergence of US-like PEDV in those countries, resulting in severe disease outbreaks, warrants serious consideration as to whether immunity induced by previously existing PEDV strains could provide protection from other exotic isolates. The results of disease outbreaks in those countries suggest that cross-protection might not be completely effective. Therefore, in Vietnam, where the emergence of US-like PEDV strains has not yet been reported, intensive surveillance control programs, including the implementation of more surveys and obtaining more isolates to sequence, should be considered. The risk of virus introduction into the country should also be determined, and control programs should be implemented accordingly.
To successfully control PEDV, intentional exposure of sows with minced intestines of PEDV-infected pigs has long been suggested [39]. However, side effects of this management protocol, including an increased percentage of mummified fetuses and risks of contamination by other pathogens, have been recognized [40]. Vaccination has been implemented as an alternative tool. The degree of successful control, however, is controversial. The route of vaccination and the genetically distinct isolates that are used to manufacture vaccines have contributed to the unsuccessful results. Because mucosal immunity, especially secretory immunoglobulin A, plays an important role in controlling PEDV infection [40], intramuscular vaccination with PEDV vaccine might not be completely effective. In addition, genetic differences between the available vaccines and the isolates responsible for outbreaks are of concern. Several studies from China have reported severe PED outbreaks even though vaccination had been extensively used in the herd [41]. However, the isolates responsible for outbreaks in China from 2011 to 2012 were new variants. The vaccines available are manufactured from isolates known as classical variants, which are genetically distinct from the new variants. Thus, vaccines manufactured from classical variants may not provide an adequate solution for successfully controlling PEDV in Vietnam. A vaccine manufactured from new-variant PEDV in conjunction with the appropriate route of administration could be used to successfully control PED in Vietnam.
In conclusion, the results of the present study demonstrate that PEDV isolates from Vietnam are new variants, closely related to Chinese PEDV and possessing a characteristic in the spike gene defined by two insertions and one deletion marker that are genetically the same as those of isolates responsible for the outbreaks from 2010 to 2012 in China. This finding suggests that vaccination based on old seed stock including classical variants might not result in successful PED control. While a new generation of vaccines based on new-variant isolates are being developed, planned exposure with PEDV-infected intestines might be an effective prevention strategy in spite of the risk of side effects. This is the first report of the entire genome of PEDV in Vietnam. We hope that these data will provide a better understanding of the molecular epidemiology and genetic diversity of PEDV field isolates in Vietnam and will help to improve the prevention strategy for controlling the disease in the future.
References
Debouck P, Pensaert M (1980) Experimental infection of pigs with a new porcine enteric coronavirus, CV 777. Am J Vet Res 41:219–223
Pensaert M, De Bouck P (1978) A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247
Pospischil A, Stuedli A, Kiupel M (2002) Update on porcine epidemic diarrhea. J Swine Health Prod 10:81–85
Chasey D, Cartwright S (1978) Virus-like particles associated with porcine epidemic diarrhoea. Res Vet Sci 25:255
Kocherhans R, Bridgen A, Ackermann M, Tobler K (2001) Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus genes 23:137–144
Song D, Park B (2012) Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus genes 44:167–175
Godet M, Grosclaude J, Delmas B, Laude H (1994) Major receptor-binding and neutralization determinants are located within the same domain of the transmissible gastroenteritis virus (coronavirus) spike protein. J Virol 68:8008–8016
Li BX, Ge JW, Li YJ (2007) Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology 365:166–172
Spaan W, Cavanagh D, Horzinek M (1988) Coronaviruses: structure and genome expression. J Gen Virol 69:2939–2952
Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, Laude H, Yang MS, Jang YS (2002) Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells 14:295–299
Cruz DJ, Kim CJ, Shin HJ (2006) Phage-displayed peptides having antigenic similarities with porcine epidemic diarrhea virus (PEDV) neutralizing epitopes. Virology 354:28–34
Cruz DJ, Kim CJ, Shin HJ (2008) The GPRLQPY motif located at the carboxy-terminal of the spike protein induces antibodies that neutralize Porcine epidemic diarrhea virus. Virus Res 132:192–196
Sun D, Feng L, Shi H, Chen J, Cui X, Chen H, Liu S, Tong Y, Wang Y, Tong G (2008) Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet Microbiol 131:73–81
Lee DK, Park CK, Kim SH, Lee C (2010) Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Res 149:175–182
Li ZL, Zhu L, Ma JY, Zhou QF, Song YH, Sun BL, Chen RA, Xie QM, Bee YZ (2012) Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field strains in south China. Virus genes 45:181–185
Park SJ, Moon HJ, Yang JS, Lee CS, Song DS, Kang BK, Park BK (2007) Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus genes 35:321–332
Park SJ, Kim HK, Song DS, Moon HJ, Park BK (2011) Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field isolates in Korea. Arch Virol 156:577–585
Park SJ, Song DS, Ha GW, Park BK (2007) Cloning and further sequence analysis of the spike gene of attenuated porcine epidemic diarrhea virus DR13. Virus genes 35:55–64
Park SJ, Moon HJ, Luo Y, Kim HK, Kim EM, Yang JS, Song DS, Kang BK, Lee CS, Park BK (2008) Cloning and further sequence analysis of the ORF3 gene of wild- and attenuated-type porcine epidemic diarrhea viruses. Virus genes 36:95–104
Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66:193–292
Zhang Z, Chen J, Shi H, Chen X, Shi D, Feng L, Yang B (2012) Identification of a conserved linear B-cell epitope in the M protein of porcine epidemic diarrhea virus. J Virol 9:225
Egberink HF, Ederveen J, Callebaut P, Horzinek MC (1988) Characterization of the structural proteins of porcine epizootic diarrhea virus, strain CV777. AmJ Vet Res 49:1320–1324
Lai MM (1990) Coronavirus: organization, replication and expression of genome. Annu Rev Microbiol 44:303–333
Schelle B, Karl N, Ludewig B, Siddell SG, Thiel V (2005) Selective replication of coronavirus genomes that express nucleocapsid protein. J Virol 79:6620–6630
Takahashi K, Okada K, Ohshima K (1983) An outbreak of swine diarrhea of a new-type associated with coronavirus-like particles in Japan. Jpn J Vet Sci 45:829–832
Chen JF, Sun DB, Wang CB, Shi HY, Cui XC, Liu SW, Qiu HJ, Feng L (2008) Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes 36:355–364
Temeeyasen G, Srijangwad A, Tripipat T, Tipsombatboon P, Piriyapongsa J, Phoolcharoen W, Chuanasa T, Tantituvanont A, Nilubol D (2013) Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infect Genet Evol 21c:205–213
Duy DT, Toan NT, Puranaveja S, Thanawongnuwech R (2013) Genetic characterization of porcine epidemic diarrhea virus (PEDV) isolates from southern Vietnam during 2009-2010 outbreaks. Thai J Vet Med 41:55–64
Hofmann M, Wyler R (1988) Propagation of the virus of porcine epidemic diarrhea in cell culture. J Clin Microbiol 26:2235–2239
Pan Y, Tian X, Li W, Zhou Q, Wang D, Bi Y, Chen F, Song Y (2012) Isolation and characterization of a variant porcine epidemic diarrhea virus in China. J Virol 9:195
Sambrook J, Russell DW (2006) Rapid Amplification of 5′ cDNA Ends (5′-RACE). Cold Spring Harbor Protoc. 2006:pdb.prot3989
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22:4673–4680
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Bio Evol 28:2731–2739
Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q (2012) New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18:1350–1353
Song D, Huang D, Peng Q, Huang T, Chen Y, Zhang T, Nie X, He H, Wang P, Liu Q, Tang Y (2015) Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea viruses associated with outbreaks of severe diarrhea in piglets in jiangxi, china 2013. PloS One 10:e0120310
Yang DQ, Ge FF, Ju HB, Wang J, Liu J, Ning K, Liu PH, Zhou JP, Sun QY (2014) Whole-genome analysis of porcine epidemic diarrhea virus (PEDV) from eastern China. Arch Virol 159:2777–2785
Choi JC, Lee KK, Pi JH, Park SY, Song CS, Choi IS, Lee JB, Lee DH, Lee SW (2014) Comparative genome analysis and molecular epidemiology of the reemerging porcine epidemic diarrhea virus strains isolated in Korea. Infect Genet Evol 26:348–351
Lin CN, Chung WB, Chang SW, Wen CC, Liu H, Chien CH, Chiou MT (2014) US-like strain of porcine epidemic diarrhea virus outbreaks in Taiwan, 2013-2014. J Vet Med Sci 76:1297–1299
Nilubol D, Khatiworavage C (2012) Control of porcine epidemic diarrhea virus through intestinal feedback and parity segregation system: field cases. In: IPVS, Jeju, Korea, p 222
Nilubol D, Khatiworavage C (2012) Comparitive efficacy of oral administration of minced piglet intestine vs intramusculor vaccination in the control porcine epidemic diarrhea in gilts. In: IPVS, Jeju, Korea, p 381
Sun RQ, Cai RJ, Chen YQ, Liang PS, Chen DK, Song CX (2012) Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis 18:161–163
Acknowledgments
We gratefully acknowledge the graduate scholarship program of Chulalongkorn University and faculty members from neighboring countries. This study was supported by the National Research Council of Thailand and the Ratchadaphiseksomphot Endowment Fund 2013 of Chulalongkorn University (CU-56-527-HR).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2015_2463_MOESM2_ESM.pdf
Supplementary Material 2 Multiple sequence alignments of the spike protein of porcine epidemic diarrhea virus identified in Vietnam and other countries. The spike protein of three Vietnamese PEDV isolates possesses two insertions of four (56GENQ59) and one (140N) amino acids at positions 56-59 and 140, respectively, and one deletion of two amino acids (160DG161) at positions 160-161 (red lines). The insertion and deletion markers are located in the hypervariable domain of the N-terminus of the S1 region. (pdf 335 kb)
Rights and permissions
About this article

Cite this article
Vui, D.T., Thanh, T.L., Tung, N. et al. Complete genome characterization of porcine epidemic diarrhea virus in Vietnam. Arch Virol 160, 1931–1938 (2015). https://doi.org/10.1007/s00705-015-2463-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2463-6