
Abstract
Among other Lyssaviruses, Daubenton’s and pond-bat-related European bat lyssavirus type 2 (EBLV-2) can cause human rabies. To investigate the diversity and evolutionary trends of EBLV-2, complete genome sequences of two Finnish isolates were analysed. One originated from a human case in 1985, and the other originated from a bat in 2009. The overall nucleotide and deduced amino acid sequence identity of the two Finnish isolates were high, as well as the similarity to fully sequenced EBLV-2 strains originating from the UK and the Netherlands. In phylogenetic analysis, the EBLV-2 strains formed a monophyletic group that was separate from other bat-type lyssaviruses, with significant support. EBLV-2 shared the most recent common ancestry with Bokeloh bat lyssavirus (BBLV) and Khujan virus (KHUV). EBLV-2 showed limited diversity compared to RABV and appears to be well adapted to its host bat species. The slow tempo of viral evolution was evident in the estimations of divergence times for EBLV-2: the current diversity was estimated to have built up during the last 2000 years, and EBLV-2 diverged from KHUV about 8000 years ago. In a phylogenetic tree of partial N gene sequences, the Finnish EBLV-2 strains clustered with strains from Central Europe, supporting the hypothesis that EBLV-2 circulating in Finland might have a Central European origin. The Finnish EBLV-2 strains and a Swiss strain were estimated to have diverged from other EBLV-2 strains during the last 1000 years, and the two Finnish strains appear to have evolved from a common ancestor during the last 200 years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rabies is a fatal and incurable zoonotic disease caused by RNA viruses belonging to the genus Lyssavirus within the family Rhabdoviridae. The negative-sense lyssavirus genome encodes five proteins: nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G) and RNA polymerase (L) in the order 3′-N-P-M-G-L-5′. Phylogenetic analysis and the virus-host relationship suggest that all 15 currently known lyssaviruses probably originated in bats and can be divided into phylogroups. Phylogroup I comprises the classical rabies virus (RABV) and the majority of bat lyssaviruses, whereas Lagos bat virus (LBV) and Shimoni bat virus (SHIBV) form phylogroup II [1, 2]. West Caucasian bat virus (WCBV), Ikoma lyssavirus (IKOV) and Lleida bat lyssavirus (LLEBV) may be representatives of a possible new phylogroups [3–5].
The first record of a rabid bat in Europe occurred in Germany in 1954 [6], but only with the advent of antigenic typing and molecular tools could European bat lyssavirus (EBLV) be characterised and distinguished from classical RABV and other lyssaviruses [7, 8]. After the death of a Swiss bat biologist in Finland in 1985 and subsequent characterisation of the causative agent, it became evident that bat rabies in Europe at the time was caused by two different lyssaviruses, EBLV-1 and 2 [7, 9]. Both of these viruses are genetically and antigenically related to RABV but are significantly different from each other. Early genomic sequencing also indicated that two distinct genetic lineages (a and b) of EBLVs have evolved, which appeared to cluster geographically [10, 11].
During 1977–2013, 1064 rabies cases were reported in 11 of the 45 known indigenous bat species in 16 European countries [12]. Although numerous human contacts with European bats, primarily from handling sick or injured animals, have been reported [13], only a few EBLV-induced human casualties have been conclusively demonstrated: in Voroshilovgrad, Ukraine (1977), in Belgorod, Russia (1985), in Finland (1985) and in the UK in Scotland (2002) [9, 14–17]. The Russian case was genetically typed as being EBLV-1a, while the Ukrainian case is only assumed to be EBLV-1 from antigenic profiling [15, 18]. The virus variants responsible for human rabies cases in the UK and Finland were typed as being EBLV-2a and b, respectively [17]. Subsequently, two other human rabies cases after a bat bite have been described in Ukraine [15]. However, these cases were diagnosed clinically and on the basis of anamnesis, and no virological or pathological results are available.
EBLV-2 is known to associate with two closely related Myotis bat species, Daubenton’s (Myotis daubentonii) and pond bats (M. dascyneme). The first isolation of EBLV-2 from a bat was carried out in 1987 from a pond bat in the Netherlands [19]. Elsewhere in Europe, EBLV-2 has been isolated sporadically from Daubenton’s bats in Switzerland [20], the UK [21, 22], Germany [23] and Finland [24]. In 1986, EBLV was detected in one Danish Daubenton’s bat and in one pond bat, but the causative strains were not analysed further. EBLV-2 was, however, demonstrated later in 2013 in mouth swabs from two Danish Daubenton’s bats using a molecular diagnostic strategy [25]. The strains from the UK and from the Netherlands appear to be closely related, as are the strains from Germany and those isolated from Switzerland [26]. The Finnish EBLV-2 strains and a strain from Switzerland have been suggested to form a lineage designated as EBLV-2b, with rest of the isolates forming the lineage EBLV-2a [10, 11]. EBLV-2 strains appear to form clusters according to the geographical area and year of isolation.
Evolutionary studies on lyssaviruses have tended to focus on the N protein, a conserved structural protein, and the G protein, which contains domains responsible for host-cell receptor recognition and membrane fusion, and which is the major target for the host neutralizing antibody response [1]. Previously, comprehensive evolutionary analysis has demonstrated that EBLV-1 and EBLV-2 have differing population structures and dispersal patterns. Molecular-clock estimates have suggested that the current linage of EBLV-1 arose some 500 to 750 years ago [27]. Little is known about the forces shaping the evolution of EBLV-2, and only three strains of EBLV-2 have previously been fully sequenced (EU293114, EF157977 and KF155004).
Finland has been free of rabies in terrestrial mammals since 1991. Nevertheless, rabies can remain a residual risk to public health due to the natural circulation of EBLV-2 [24, 28]. Here, we describe the phylogenetic analysis of two Finnish EBLV-2 strains, the first isolated from a human in 1985 and the second from a diseased bat in 2009. Using these isolates as a calibrator, this study provides an estimate of the EBLV-2 molecular clock. Increased information on the genome sequences of lyssaviruses is fundamental to understanding their epidemiology and evolution, and to demonstrating the importance of continuous surveillance and molecular characterization of lyssaviruses circulating in animal populations.
Materials and methods
Virus isolation in MNA cells and in suckling mice
Ethical approval and permission
The National Animal Experiment Board of the County Administrative Board of Southern Finland approved the diagnostic mouse inoculation test (permission number ESLH-2008-06899/Ym-23), which followed Finnish legislation, namely, the Finnish Act on the Use of Animals for Experimental Purposes (62/2006).
Human bat virus isolate FI-85 [9, 16] used in this study was the first archived newborn-mouse passage and had been kept at -70 °C since 1986. Virus isolation of the bat virus isolate FI-09 [24] was attempted in MNA cell culture (mouse neuroblast cells, Neuro-2a [ATCC® CCL-131™]), with three additional passages according to a procedure described in the OIE manual [29]. The bat brain suspension was subsequently inoculated into suckling mice. Eight newborn mice (ScaNmri) at 2 days old were intracerebrally infected with 20 µl of the suspension in a BSL-3 laboratory. When the mice started to develop signs of encephalitis, they were euthanized and the brains were collected and examined using a fluorescence antibody test (FAT) for the presence of lyssavirus as described earlier by Jakava-Viljanen et al. [24].
RNA extraction, primer design and RT-PCR
RNA was extracted from the brain suspensions of the mice using a QIAamp Viral RNA Mini Kit (QIAGEN, Germany, Hilden) according to the manufacturer’s instructions. Primers (Table 1) were designed with the program PCR Suite [30] and modified afterwards, if needed. A OneStep RT-PCR Kit (QIAGEN, Hilden, Germany) was used to amplify 34 overlapping fragments. The following thermal profile was used: a single cycle of reverse transcription for 30 min at 50 °C, 15 min at 94 °C for reverse transcriptase inactivation and DNA polymerase activation, followed by 30 amplification cycles of 1 min at 94 °C, 1 min at 50 °C and 1 min at 72 °C. After agarose gel electrophoresis, the bands were cut from the gel and DNA was extracted using a QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany).
Sequencing and sequence analysis
PCR products were sequenced using an ABI 3100 Avant Genetic Analyser (Applied Biosystems) with the primers used in the PCR and a Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Before sequencing, the reaction products were purified using a DyeEx 2.0 Spin Kit (QIAGEN, Hilden Germany). The sequences were analysed with Sequencing Analysis software version 6.0 (Applied Biosystems).
Phylogenetic analysis
The newly determined sequences (GenBank accession numbers JX129232 and JX129233) were aligned with other published full-length lyssavirus sequences from bat-type isolates obtained from GenBank to evaluate the genetic diversity of EBLV-2 strains and their relationship to other bat-associated genotypes. Amino acid sequences were deduced using the “translate” function of the program MEGA v.5.1 [31]. Phylogenetic trees of full-length genomes and partial N coding sequences were calculated using the maximum-likelihood approach in the program MEGA with 1000 bootstrap replicates. The pairwise sequence identities were calculated using CLUSTAL W2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/), with default settings.
Rate of evolution and time of divergence
The program BEAST [32] was used to estimate the substitution rate of EBLV-2 and time of divergence from the phylogenetically closest lyssaviruses. The data set used for estimating the rate of evolution was based on the partial N coding sequences (400 bp) of EBLV-2 strains isolated from the years 1986 to 2012. The HKY model of nucleotide substitution was used [33] with both strict and relaxed lognormal clock models. Convergence of parameters was assessed using TRACER [32], and each run was continued until the effective sampling size of all parameters was greater than 200.
Results
Isolation of the EBLV-2 FI-09 strain in MNA cells and suckling mice
Inoculation of the bat sample into MNA cells revealed the presence of virus in only a limited number of cells after 3 days of incubation, and after the three subsequent passages, the tissue culture infection test was negative. When the cell culture isolation was unsuccessful, the brain suspension of the bat was inoculated into suckling mice. The mice began to develop signs of encephalitis on day 12 p.i. By day 17 p.i., all eight mice were euthanized. The presence of the virus in all of the brain samples was confirmed by FAT.
EBLV-2 genome, genomic distance and phylogenomic pattern
The complete lengths of the FI-85 and FI-09 genomes were 11,928 and 11,927 nucleotides, respectively (GenBank accession numbers JX129233-JX129232). Twenty nucleotides at the 5’ end and 22 nucleotides at the 3’ end of the genomes were primer-derived and were excluded from all phylogenetic analyses.
The general genome organization was typical for lyssaviruses, consisting of five structural genes, N, P, M, G and L, and non-coding regions between them and at both ends of the genome. The lengths of the genes and the number of deduced amino acids (in parentheses) were 1356 (451 aa), 894 (297 aa), 609 (202 aa), 1575 (524 aa) and 6384 (2127 aa), respectively. The long untranslated region between the G and L genes was the only area that varied in length between the two Finnish EBLV-2 strains, being 510 bp in FI-85 and 509 bp in FI-09. The corresponding lengths for other completely sequenced EBLV-2 strains were 511 bp for EU293114 and 512 bp for KF155004 and EF157977.
The nucleotide sequence identity between the FI-85 and FI-09 strains was 99.6 %. There were four amino acid differences between the two Finnish strains: N gene aa 106 (N><S), G gene aa 158 (V><A), L gene aa 154 (R><K) and aa 1656 (G><D). The N, P, M, G and L genes of the FI-85 and FI-09 strains shared 98–99 %, 98 %, 99–100 %, 97–99 % and 99 % amino acid sequence identity, respectively, with the previously published EBLV-2 strains. The similarity plot in Figure 1 displays the similarities along the genome. The non-coding regions are highly divergent, yet rather similar for all EBLV-2 strains.

Similarity plot of bat-related lyssavirus genomes. Similarity is shown on the y-axis, and the position in the genome is shown on the x-axis
Phylogenetic analysis and evolutionary trends
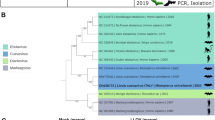
A phylogenetic tree was constructed based on the genome sequences of the two new EBLV-2 isolates and the full-length genome sequences of bat-related lyssavirus obtained from GenBank (Fig. 2). The analysis revealed that all five EBLV-2 strains are monophyletic. The phylogenetic tree also demonstrated that EBLV-2 strains share the most recent common ancestry with BBLV and KHUV, and more distant ancestry with ARAV, RABV and ABLV (albeit with low bootstrap support). A second major cluster of lyssaviruses consists of IRKV, DUVV and EBLV-1. The clearly most divergent group is formed by SHIV, LBV and WCBV.

Phylogenetic tree of lyssaviruses estimated using complete coding sequences. The phylogenetic tree was calculated using the maximum-likelihood approach in the program MEGA [30] with 1000 bootstrap replicates
In order to include more EBLV-2 strains, we constructed a second phylogenetic tree (Fig. 3) based on partial N gene sequences (400 nt), which are much more abundant in the GenBank database. EBLV-2 strains were also monophyletic in this region. Some discrepancies were observed between the two trees, especially in the composition of the major clusters, which were not reproduced in the phylogeny based on partial N gene sequences.

Phylogenetic tree based on partial N gene (400 nt) sequences. The phylogenetic tree was calculated using the maximum-likelihood approach in the program MEGA [31] with 1000 bootstrap replicates
The substitution rate of European bat lyssavirus-2 was estimated using the two Finnish strains isolated 24 years apart as a calibrator. The overall rate was 7.67 × 10−5 substitutions per site per year. The current diversity of EBLV-2 was estimated to have appeared during the last 2000 years. However, EBLV-2 and other phylogroup I viruses were estimated to have already diverged from other lyssaviruses at about 8000 years ago (Fig. 4). The divergence of Finnish EBLV-2 strains and a strain from Switzerland from other EBLV-2 strains has occurred during the last 1000 years. The two Finnish strains have evolved from a common ancestor during the last 200 years.

Phylogenetic tree of EBLV-2 based on partial N gene (400 nt) sequences with a molecular-clock estimate. The program BEAST [32] was used to estimate the substitution rate of EBLV-2 and time of divergence from the phylogenetically closest lyssavirus. The HKY model of nucleotide substitution was used [33] with both strict and relaxed lognormal clock models. Convergence of parameters was assessed using TRACER [31], and each run was continued until the effective sampling size of all parameters was greater than 200
Discussion
Sequencing, phylogenetic analysis and evolutionary analysis of Finnish EBLV-2 strains isolated from a human case in 1985 and from a bat in 2009 were described. It was shown that the EBLV-2 isolates share high nucleotide and amino acid sequence identity, regardless of the year and origin of isolation, but they also cluster according to the host species and the geographical place of isolation.
The closest relatives to EBLV-2 (Bokeloh and KHUV) are lyssavirus strains from Myotis bats from Germany and Tajikistan. An interesting case is ARAV (originating from Kyrgystan), which has an uncertain position in the phylogenetic analysis. The possibility of recombination of rabies viruses has been demonstrated in earlier studies [34–36]. Lleida bat lyssavirus, detected in a bent-winged bat (M. schreibersii) in Spain in 2011 [5], is the most unique lyssavirus in Europe based on the N gene, although the whole genome sequence is not available. WCBLV has also been detected in M. schreibersii, and antibodies against WCBV have been detected in Africa in Miniopterus bats [37]. The tree topology was somewhat different in the phylogeny based on either the complete coding sequences or the commonly used partial N sequences, and it should be appreciated that for a detailed analysis of evolutionary history, complete genomes should be used. The limitation of this study, as in other studies, is the amount of data available for comparison.
The nucleotide sequence identity (deduced amino acid sequence identity) between FI-85 and FI-09 was 99.6 % (99.8 %). The N, M and L proteins were similar in structure and length between members of different lyssavirus species and strains, whereas the lengths of the P and cytoplasmic regions of the G protein were variable [38–40]. No differences in structure were seen in the Finnish EBLV-2 isolates in comparison to other EBLV-2 complete genomes [40–42]. The similarity plot in Figure 1 further demonstrates the similarities along the genomes of different lyssaviruses. Interestingly, the non-coding regions, which are highly divergent in other lyssaviruses, were rather similar for all EBLV-2 strains.
Based on this study, we estimate that the Finnish EBLV-2 strains and one Swiss strain from among the other EBLV-2 strains have evolved from a common ancestor during the last 1000 years. Earlier, the Finnish strains and the strain from Switzerland were suggested to form the lineage EBLV-2b, whereas the rest of the isolates form the lineage EBLV-2a [10, 11, 20, 27]. In our study, there was a division into subgroups EBLV-2a and EBLV-2b when full genomes were compared (Fig. 2), but when more strains were compared using shorter, 400-bp N gene sequences, we could not confirm the suggested division into subgroups (Fig. 3). In our comparison, the Finnish strains and the strain from Switzerland also clustered together based on N gene analysis. Interpretation of the molecular epidemiology of the strains is further complicated by the history of the Swiss bat biologist who died in Finland in 1985 of EBLV-2 infection (JX129233), who had been bitten 51 days before the onset of clinical signs by a Daubenton’s bat that was abnormal. The bat was freed before the patient developed symptoms and was not available for rabies analysis [16]. However, it is possible that the bat originated from Central Europe, possibly from Switzerland, even though the exposure took place in Finland.
The molecular clock estimates suggest that the current linage of EBLV-2 arose some 2000 years ago and that the most recent common ancestor (MRCA) occurred around 8000 years ago. In earlier molecular clock studies, Tao et al. [43] estimated based on G gene analysis that the time to the most common ancestor (TMRCA) of all lyssaviruses was approximately 5030 years. The West Caucasian bat virus divided first, and then phylogroup I and phylogroup II divided about 4000 years ago [43]. Davis et al. [27] estimated that the MRCA of EBLV-1 existed around 500 to 750 years ago based on G and N sequences, or 70 to 300 years ago, depending on the substitution rate [44]. The molecular clock estimates cannot be used to precisely describe events that occurred thousands, or even tens of thousands of years ago, since sequence data only exist from the last few decades. The overall rate of evolution appears somewhat slower for the bat-type lyssaviruses than for RABV. This is in line with bats being considered the true reservoir of lyssaviruses. The overall evolution rate for EBLV-2 in our study was 7.67 × 10−5 substitutions per site per year. For RABV, it has been suggested to be 1.56–1.78 × 10−4 [45], and 1 × 10−3 to 5.5 × 10−4 [7, 43, 46, 47], and for EBLV-1, 5–6 × 10−5 to 5–6 × 10−4 [27].
Behavioural and ecological traits of bats, such as hibernation, migration and coloniality, might influence viral evolution. Some rabies virus lineages evolve up to 22 times faster than others, depending on the reservoir host [48, 49]. Virus lineages in bat species in the temperate climate zone were found to have four times slower rates of evolution than lineages in bats from tropical or subtropical climates, and viral evolution was faster in bats that remain active year-round in comparison to bats that hibernate. This could be a major factor in a cold climate region such in Finland. Viral evolutionary rates were similarly unconstrained by host evolutionary relatedness such that lyssaviruses associated with closely related bat species or subspecies often had dissimilar evolutionary rates. Streicker et al. [48] demonstrated that the local host environment determines the evolutionary rates of lyssaviruses. Daubenton’s bats are facultatively seasonal migrants covering middle-range distances of 100–150 km [50], which supports the clustering of EBLV-2 according to the geographical place of isolation. In Finland, antibodies to EBLVs have only been detected in a restricted geographical area close to the location where the EBLV-2-positive bat was found in 2009 [28]. In a study of Daubenton’s bats from Finland, the UK, Switzerland and Spain, no population structuring was observed [51]. Daubenton’s bats migrate between the UK and the mainland, and the genetic structure of Daubenton’s bats is relatively homogeneous in western parts of Europe [52]. This could explain the relatedness within the proposed lineage 2a. As the migration pattern of Daubenton’s bats does not support the finding that the Finnish EBLV-2 Daubenton’s bat strain is closely related to the EBLV-2 strain from Switzerland, one can speculate whether there could have been contact between indigenous Finnish bats and the bats that were freed in Finland and whose origin is unknown [9, 16].
Increased information on the complete genome sequences of lyssaviruses is fundamental to understanding their epidemiology and evolution and demonstrates the importance of continuous monitoring and molecular characterization of lyssaviruses circulating in human and animal populations.
References
Badrane H, Bahloul C, Perrin P, Tordo N (2001) Evidence of two Lyssavirus phylogroups with distinct pathogenicity and immunogenicity. J Virol 75:3268–3276
Kuzmin IV, Mayer AE, Niezgoda M, Markotter W, Agwanda B, Breiman RF, Rupprecht CE (2010) Shimoni bat virus, a new representative of the Lyssavirus genus. Virus Res 149:197–210
Kuzmin IV, Hughes GJ, Botvinkin AD, Orciari LA, Rupprecht CE (2005) Phylogenetic relationships of Irkut and West Caucasian bat viruses within the Lyssavirus genus and suggested quantitative criteria based on the N gene sequence for lyssavirus genotype definition. Virus Res 111:28–43
Marston DA, Horton DL, Ngeleja C, Hampson K, McElhinney LM, Banyard AC, Haydon D, Cleaveland S, Rupprecht CE, Bigambo M, Fooks AR, Lembo T (2012) Ikoma lyssavirus, highly divergent novel lyssavirus in an African civet. Emerg Infect Dis 18:664–667
Ceballos NA, Morón SV, Berciano JM, Nicolás O, López CA, Juste J, Nevado CR, Setién AA, Echevarría JE (2013) Novel lyssavirus in bat, Spain. Emerg Infect Dis 19:793–795
Mohr W (1957) Die Tollwut. Med Klinik 52:1057–1060
Bourhy H, Kissi B, Lafon M, Sacramento D, Tordo N (1992) Antigenic and molecular characterization of bat rabies virus in Europe. J Clin Microbiol 30(9):2419–2426
Schneider LG, Cox JH (1994) Bat lyssaviruses in Europe. Curr Top Microbiol Immunol 187:207–218
Lumio J, Hillbom M, Roine R, Ketonen L, Haltia M, Valle M, Neuvonen E, Lähdevirta J (1986) Human rabies of bat origin in Europe. Lancet 15:378
Amengual B, Whitby JE, King A, Cobo JS, Bourhy H (1997) Evolution of European bat lyssaviruses. J Gen Virol 78:2319–2328
McElhinney LM, Marston DA, Leech S, Freuling CM, van der Poel WH, Echevarria J, Vázquez-Moron S, Horton DL, Müller T, Fooks AR (2013) Molecular epidemiology of bat lyssaviruses in Europe. Zoonoses Public Health 60:35–45
WHO Rabies Bulletin Europe. http://www.who-rabies-bulletin.org/
Brass DA (1994) Rabies in bats. Natural History and Public Health Implications. Ridgefield CN, Livia Press
Anonymous (1986) Bat rabies in the Union of Soviet Socialist Republics. Rabies Bull Eur 4:12–14
Botvinkin AD, Poleschuk EM, Kuzmin IV, Borisova TI, Gazaryan SV, Yager P, Rupprecht CE (2003) Novel lyssaviruses isolated from bats in Russia. Emerg Infect Dis 9:1623–1625
Roine RO, Hillbom M, Valle M, Haltia M, Ketonen L, Neuvonen E, Lumio J, Lähdevirta J (1988) Fatal encephalitis caused by a bat-borne rabies-related virus. Clinical findings. Brain 111:1505–1516
Fooks AR, McElhinney LM, Pounder DJ, Finnegan CJ, Mansfield K, Johnson N, Brookes SM, Parsons G, White K, McIntyre PG, Nathwani D (2003) Case report: isolation of a European bat lyssavirus type 2a from a fatal human case of rabies encephalitis. J Med Virol 71:281–289
Selimov MA, Tatarov AG, Botvinkin AD, Klueva EV, Kulikova LG, Khismatullina NA (1989) Rabies-related Yuli virus; identification with a panel of monoclonal antibodies. Acta Virol 33:542–546
Nieuwenhuijs J (1987) WHO Rabies Bulletin Europe 4:8. http://www.who-rabies-bulletin.org/
Megali A, Yannic G, Zahno ML, Brügger D, Bertoni G, Christe P, Zanoni R (2010) Surveillance for European bat lyssavirus in Swiss bats. Arch Virol 155:1655–1662
Whitby JE, Heaton PR, Black EM, Wooldridge M, McElhinney LM, Johnstone P (2000) First isolation of a rabies-related virus from a Daubenton’s bat in the United Kingdom. Vet Rec 147:385–388
Harris SL, Mansfield K, Marston DA, Johnson N, Pajamo K, O’Brien N, Black C, McElhinney LM, Fooks AR (2007) Isolation of European bat lyssavirus type 2 from a Daubenton’s bat (Myotis daubentonii) in Shropshire. Vet Rec 161:384–386
Freuling C, Grossmann E, Conraths FJ, Schameitat A, Kliemt J, Auer E, Greiser-Wilke I, Müller T (2008) First isolation of EBLV-2 in Germany. Vet Microbiol 131:26–34
Jakava-Viljanen M, Lilley T, Kyheröinen E-M, Huovilainen A (2010) First encounter of European bat lyssavirus type 2 (EBLV-2) in a bat in Finland. Epidemiol Infect 138:1581–1585
Rasmussen TB, Chrlél M, Baagøe, Fjederholt E, Kooi EA, Belsham GJ, Bøtner A (2014) Detection of European bat lyssavirus type 2 in Danish Daubenton’s bats using a molecular diagnostic strategy. In Proceedings of 8th annual EPIZONE meeting, Copenhagen
Schatz J, Ohlendorf B, Busse P, Pelz G, Dolch D, Teubner J, Encarnação JA, Mühle RU, Fischer M, Hoffmann B, Kwasnitschka L, Balkema-Buschmann A, Mettenleiter TC, Müller T, Freuling CM (2014) Twenty years of active bat rabies surveillance in Germany: a detailed analysis and future perspectives. Epidemiol Infect 142:1155–1166. doi:10.1017/S0950268813002185
Davis PL, Holmes EC, Larrous F, Van der Poel WH, Tjørnehøj K, Alonso WJ, Bourhy H (2005) Phylogeography, population dynamics and molecular evolution of European bat lyssaviruses. J Virol 79:10487–10497
Nokireki T, Huovilainen A, Lilley T, Kyheröinen EM, Ek-Kommonen C, Sihvonen L, Jakava-Viljanen M (2013) Bat rabies surveillance in Finland. BMC Vet Res 9:174
OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2014. http://www.oie.int/en/international-standard-setting/terrestrial-manual/access-online/
van Baren P, Heutink (2004) The PCR Suite M.J. Bioinformatics 20:591–593
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 8(7):214
Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Kuzmin I, Orciari L, Arai T, Smith J, Hanlon C, Kameoka Y, Rupprecht C (2003) Bat lyssaviruses (Aravan and Khujand) from Central Asia: phylogenetic relationship according to N, P and G gene sequences. Virus Res 97:65–79
Wei L, Yanfeng L, Jing L, Jianwei Z, Youhua X (2011) Evidence for inter- and intra-clade recombinations in rabies virus. Infect Genet Evol 11:1906–1912
He C-Q, Meng S-L, Yan H-Y, Ding N-Z, He H-B et al (2012) Isolation and Identification of a novel rabies virus lineage in China with natural recombinant nucleoprotein gene. PLoS ONE 7(12):e49992. doi:10.1371/journal.pone.0049992
Kuzmin IV, Niezgoda M, Franka R, Agwanda B, Markotter W, Beagley JC, Urazova OY, Breiman RF, Rupprecht CE (2008) Possible emergence of West Caucasian bat virus in Africa. Emerg Infect Dis 14:1887–1889
Le Mercier P, Jacob Y, Tordo N (1997) The complete Mokola virus genome sequence: structure of the RNA-dependent RNA polymerase. J Gen Virol 78:1571–1576
Warrilow D, Smith IL, Harrower B, Smith GA (2002) Sequence analysis of an isolate from a fatal human infection of Australian bat lyssavirus. Virology 297:109–119
Marston DA, McElhinney LM, Johnson N, Müller T, Conzelmann KK, Tordo N, Fooks AR (2007) Comparative analysis of the full genome sequence of European bat lyssavirus type 1 and type 2 with other lyssaviruses and evidence for a conserved transcription termination and polyadenylation motif in the G-L 3′ non-translated region. J Gen Virol 88:1302–1314
Delmas O, Holmes EC, Talbi C, Larrous F, Dacheux L, Bouchier C, Bourhy H (2008) Genomic diversity and evolution of lyssaviruses. PLoS ONE 3:e2057. doi:10.1371/journal.pone.0002057
Marston DA, McElhinney LM, Ellis RJ, Horton DL, Wise EL, Leech SL, David D, de Lamballerie X, Fooks AR (2013) Next generation sequencing of viral RNA genomes. BMC Genomics 14:444
Tao X, Guo Z, Li H, Han N, Tang Q, Liang G (2013) Investigation of the evolutionary history of the lyssaviruses. Virol Sin 28:186–189
Hughes GJ (2008) A reassessment of the emergence time of European bat lyssavirus type 1. Infect Gen Evol 8:820–824. doi:10.1016/j.meegid.2008.08.003
Kuzmina NA, Kuzmin I, Ellison JA, Taylor ST, Bergman DL, Dew B, Rupprecht CE (2013) A reassessment of the evolutionary timescale of bat rabies viruses based upon glycoprotein gene sequences. Virus Genes 47:305–310
Badrane H, Tordo N (2001) Host switching in Lyssavirus history from the Chiroptera to the Carnivora orders. J Virol 75:8096–8104
Hanada K, Suzuki Y, Gojobori T (2004) A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol Biol Evol 21:1074–1080
Streicker DG, Lemey P, Velasco-Villa A, Rupprecht CE (2012) Rates of viral evolution are linked to host geography in bat rabies. PLoS Pathog 8(5):e1002720. doi:10.1371/journal.ppat.1002720
Mollentze N, Biek R, Streicker DG (2014) The role of viral evolution in rabies host shifts and emergence. Curr Opin Virol 8:68–72
Hutterer R, Ivanova T, Meyer-Cords R, Rodrigues L (2005) Bat migrations in Europe. A review of banding data and literature. Federal Agency for Nature Conservation, Bonn, p 162 (Naturshcutz und Biologische Vielfalt Heft 28)
Laine VN, Lilley TM, Norrdahl K, Primmer CR (2013) Population genetics of Daubenton’s bat (Myotis daubentonii) in the Archipelago Sea, southwest Finland. Ann Zool Fenn 50:303–315
Smith GC, Aegerter JN, Allnutt TR, MacNicoll AD, Learmount J, Hutson AM, Atterby H (2011) Bat population genetics and Lyssavirus presence in Great Britain. Epidemiol Infect 10:1463–1469
Acknowledgments
Special thanks to Tiina Peltonen and other technicians at Evira for technical assistance. This study was funded by the Ministry of Agriculture and Forestry (grant MMM 2682/311/2009) and the Finnish Food Safety Authority Evira.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
The National Animal Experiment Board of the County Administrative Board of Southern Finland approved the diagnostic mouse inoculation test (permission number ESLH-2008-06899/Ym-23), which followed Finnish legislation, namely the Finnish Act on the Use of Animals for Experimental Purposes (62/2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Miia, JV., Tiina, N., Tarja, S. et al. Evolutionary trends of European bat lyssavirus type 2 including genetic characterization of Finnish strains of human and bat origin 24 years apart. Arch Virol 160, 1489–1498 (2015). https://doi.org/10.1007/s00705-015-2424-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2424-0