
Abstract
Human metapneumovirus (hMPV) is associated with respiratory infections in both adults and children. Recombinant nucleocapsid (N) proteins of hMPV from two major groups of hMPV in Beijing with a 6× His-tag at the C terminus were constructed in a baculovirus and expressed in transfected Sf21 insect cells. The antigenicity and specificity of the expressed recombinant proteins were examined by immunofluorescence assay and western blotting. Preliminary use of the expressed proteins for antibody detection in 187 serum specimens collected from different age groups in Beijing, China, indicated that the purified recombinant N-His proteins were of good antigenicity and specificity and could be a potential antigen for further seroprevalence study of hMPV, especially in the Chinese population. The positive rate for antibody detection suggested that hMPV from two clusters was co-circulating in Beijing and that most of the people in Beijing have been exposed to the virus by age 60.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human metapneumovirus (hMPV) was first isolated in the Netherlands in 2001 [1] and has been classified as a member of the subfamily Pneumovirinae of the family Paramyxoviridae. It is a nonsegmented, negative-stranded RNA virus associated with respiratory tract diseases in both children and adults [2]. Very young or premature infants, patients with underlying cardiac or pulmonary disease, and immunocompromised patients are at risk for severe disease due to hMPV infection [3–5]. It was found that the virus has been circulating for at least 50 years [2], and there are two genetic clusters of hMPV that co-circulate worldwide, as shown by genetic analysis [6, 7].
It was found that hMPV with both clusters were associated with acute respiratory infections in children in Beijing, China [8]; however, the prevalence of this virus in Beijing, which could be reflected by serum antibodies against the virus, was not clear. Although hMPV has been isolated by cell culture, the virus cannot, or only poorly, be propagated in most cell lines used for isolation of respiratory viruses, such as Vero, MDCK and A-549 cells, and needs two weeks at least to display cytopathic effects in tertiary monkey kidney (tMK) or LLC-MK2 cells [1, 9]. Therefore, immunoassays for antibody detection based on virus-infected cells are ineffective. Reverse transcription polymerase chain reaction (RT-PCR) to detect viral RNA in clinical samples [10] and western blot assay or enzyme-linked immunosorbent assay (ELISA) using one or several viral proteins [11–13] have been developed to detect hMPV infections. However, there are some shortcomings to these methods, such as false positive or negative results in RT-PCR, defective post-translational modification in some expression systems, and difficulty in assessing the quality and quantity of expressed proteins in cell lysates because of differences in cell growth conditions at different times. The nucleocapsid protein (N protein) of hMPV is a major immunogen of the virus [14] and is highly conserved, with amino acid identities of 98.3–100% and 94.2–95.9% within and between genetic clusters, respectively [6, 15]. Moreover, it has been shown that the N protein is able to stimulate a substantial immune response in infected animals [16]. In this study, N-protein-encoding genes of hMPV that were amplified from clinical samples collected from children in Beijing and fused with a 6× his-tag were cloned in baculovirus and expressed in Sf21 cells. These could be easily separated by affinity chromatography to produce purified N protein to be used as antigen for immunoassays to identify serum antibody against hMPV in the Beijing population.
Materials and methods
Cloning, expression, and purification of the N protein of hMPV
Full-length cDNAs of the genes encoding the N proteins of hMPV BJ1816 and BJ1887 (GenBank accession no. DQ843658 and DQ843659), which represented two different gene clusters of hMPV circulating in Beijing, were cloned in TA vectors [15] and amplified by PCR with the following primers, which were designed based on the sequences of the N proteins from hMPV BJ1816 and BJ1887: 5′-CCGCTCGAGATGTCTCTTCAAGGG-3′ (forward), 5′-ATGGTGATGGTGATGCTCATAATCATCTTGAT-3′ (reverse) and 5′-GGGGTACCTAGTGATGGTGATGGTGATG-3′ (his-tag primer). They were then each inserted into the pFastBac1 vector. The restriction sites in the primers used for cloning purposes are underlined, and a 6× his-tag was added in the C terminus of N gene. Figure 1 shows the strategy for amplification and cloning of the hMPV N gene in the pFastBac1 vector. At first, we tried to add a 6× his-tag onto the N terminus of the N gene.

Strategy for amplifying and cloning the hMPV N gene. The constructed BJ1816/1887 N TA vector was used as a template for recombinant DNA synthesis by PCR amplification using high-fidelity Taq DNA polymerase (TaKaRa Biotechnology (Dalian) Co., Ltd)
The recombinant N BJ1816 -His and N BJ1887 -His baculoviruses were generated using transposon-mediated insertion of the corresponding genetic construct under the polyhedron promoter of Autographa californica nuclear polyhedrosis virus [17], and these constructs were introduced into competent Escherichia coli DH10BAC cells. Then, Spodopera frugiperda (Sf) 21 cells were transfected with the recombinant N BJ1816 -His baculovirus and N BJ1887 -His baculovirus, following the manufacturer’s instructions (BAC-TO-BAC baculovirus expression system, Life Technologies, USA). Mock-infected Sf21 cells and Sf21 cells infected with recombinant baculovirus obtained from pFastbac1 without any gene insertion were used as controls.
Expression of the N protein was detected by western blot using transfected Sf21 cells collected at 24, 48 and 72 h post-transfection. After the best time for expression of the recombinant protein was determined, supernatant from the infected cells was collected at 72 h post-transfection for further analysis.
The N BJ1816 -His and N BJ1887 -His proteins were purified using nickel–nitrilotriacetic acid (Ni–NTA) agarose (Qiagen, Valencia, CA) as recommended by the manufacturer (Handbook for High-Level Expression and Purification of 6xHis-Tagged Proteins, Qiagen, Valencia, CA). Briefly, the density of Sf21 cells was about 1.5 × 106/ml in shaker flasks, and the multiplicity of infection (MOI) was about 2–3. The Sf21 cells were incubated with shaking for 72 h at 27°C and harvested by centrifugation at 500×g for 5 min at 4°C. The pellet was resuspended in lysis buffer [50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole (pH adjusted to 8.0 by with NaOH), 1 mM phenylmethylsulfonyl fluoride, 1% Triton X-100]. After incubation for 30 min on ice and ultrasonic treatment at 90 Hz for 5 min, the preparation was clarified by centrifugation at 11,300 g for 15 min. The supernatant was loaded onto a Ni–NTA column that had been equilibrated with lysis buffer. The column was washed successively with ten column volumes of wash buffer [50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole (pH adjusted to 8.0 with NaOH), 1 mM phenylmethylsulfonyl fluoride]. Protein was eluted from the column with elution buffer [50 mM NaH2PO4, 300 mM NaCl, 200 mM imidazole (pH adjusted to 8.0 with NaOH)]. The samples were collected in 0.5-ml fractions. The concentration of the protein after purification was determined by the Bradford method using bovine serum albumin (Sigma, USA) as the standard.
Identification of the expressed hMPV N protein
Sf 21 cells infected with recombinant N BJ1816 -His or N BJ1887 -His baculovirus were centrifuged at 72 h post-transfection, followed by washing with phosphate-buffered saline (PBS) before being smeared onto slides (hMPV slide) and used for detecting the expression of hMPV N protein by immunofluorescent assay. Mock-infected Sf21 cells and Sf21 cells infected with recombinant baculovirus from pFastbac1 without any gene insertion were smeared onto slides and used as controls. Mouse anti-hMPV N monoclonal antibody (Chemicon International, Temecula, CA) at a dilution of 1:200 was incubated with the cell smears for 30 min at 37°C. After washing with PBS, they were incubated for 30 min at 37°C with fluorescein-isothiocyanate-conjugated goat anti-mouse IgG (Chemicon International, Temecula, CA) with 0.02% Evans blue. They were then washed twice for 5 min in PBS, air dried, and mounted with mounting medium. The hMPV slide containing control and recombinant N BJ1816 -His or N BJ1887 -His baculovirus-infected Sf21 cells and a slide with RSV- and parainfluenza virus 2 and 3-infected cells (Diagnostic Hybrids, Athens, OH, USA) were used for testing cross-reaction among hMPV, RSV and parainfluenza virus, which are all members of the family Paramyxoviridae, using fluorescein-isothiocyanate-conjugated goat polyclonal antibodies against RSV and parainfluenza virus 2/3 (Meridian Life Science, USA). Then, the slides were examined using fluorescent microscopy (Nikon E600, Japan).
Lysates from control and recombinant N BJ1816 -His or N BJ1887 -His baculovirus-infected cells were fractionated by 12% SDS polyacrylamide gel electrophoresis (PAGE) and visualized by Coomassie blue staining [18]. Rabbit anti-hMPV N hyperimmune serum (1:2,000 dilution, kindly provided by Professor Yan Li, Canada) and mouse anti-His monoclonal antibody (1:2,000 dilution, Tiangen Biotech Co, Ltd, Beijing, China) were used as the primary antibodies in western blot analysis after the proteins were electrotransferred onto the nitrocellulose membrane. Corresponding species-specific anti-immunoglobulin conjugated to horseradish peroxidase (HRP, 1:2,500 dilution, Life Technologies Corporation, Beijing, China) were used as the secondary antibodies.
Serum specimens
In total, 188 serum specimens were used in this study, including one serum specimen from a hospitalized child with pneumonia, which was used in a preliminary experiment. In February 2009, this child was diagnosed as having an hMPV infection by RT-PCR using nasopharyngial aspirates and by IFA in LLC-MK2 cells to test for hMPV-specific IgM. The other 187 serum specimens used in this study included 139 serum specimens collected from adults aged from 18 years to over 60 years who were undergoing regular health checkups at a clinic in Beijing in August 2005, and 48 serum specimens were collected from infants and children who visited the outpatient department at the Children’s Hospital Affiliated to Capital Institute of Pediatrics in Beijing in 2005 due to various illnesses. All of the serum samples were stored at −20°C before use.
Western blotting for detection of antibody against the N protein of hMPV in human sera from Beijing
The purified N protein was separated by SDS-PAGE using a 12% polyacrylamide gel and electrotransferred onto a nitrocellulose membrane. After the membrane was sliced into strips and blocked with 5% non-fat milk in PBS, sera at a dilution of 1:200 were allowed to bind to the strips, which were incubated with horseradish-peroxidase-conjugated goat anti-human IgG at dilution of 1:2,000 (Jackson ImmunoReserch Laboratories, Inc., USA), and hMPV-specific IgG from human sera binding to the N protein was detected using 3,3′-diaminobenzidine (DAB, AMRESCO Inc., USA).
ELISA for detecting antibody against the N protein of hMPV in human sera
All washing steps were carried out three times with PBST (0.05% Tween 20 in PBS). All antigens and antibodies were diluted in PBS containing 1% non-fat milk. Flat-bottom polystyrene microtiter plates (Corning Costar, USA) were used as solid-phase adsorbents. The purified recombinant N protein in PBS (0.5 μg in 100 μl/well) was added to the wells. After incubating for 18 h at 4°C, the plates were washed and then blocked with 200 μl of 5% non-fat milk in PBST for 2 h at 37°C. Subsequently, the plates were washed and incubated for 2 h at 37°C with human sera (diluted 1:200). After washing, the appropriate dilution (1:16,000) of horseradish peroxidase (HRP)-conjugated goat anti-human IgG (Jackson ImmunoResearch Laboratories, Inc., USA) was added and the plates were incubated further for 1 h at 37°C. After further washing, an enzyme substrate solution containing 0.4 mg/ml OPD (Sigma, USA) in 0.1 M citrate buffer (pH 5.0–5.5) was added. The reaction was stopped after incubation for 15 min at room temperature, and the A490 values were measured with a microtiter plate reader (Molecular Devices Corp., USA). Forty-five out of these 188 serum samples that were negative in western blot (both for 1816N and 1887N as antigen) were selected as negative samples. The average OD 490 nm value for the samples was 0.219, and the cutoff value was set at 0.46 (OD negative × 2.1).
Statistical analysis
The data were analyzed by chi-square test (χ2 test) and consistency checking (Kappa value test). P < 0.05 is the level for significant difference. A kappa value >0.75 represents very good consistency, >0.4 represents acceptable consistency.
Results
Expression and purification of the recombinant his-tagged N protein
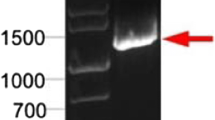
Sf21 cells transfected with recombinant baculovirus generated at first with a 6× his-tag added at N terminus of the N gene for both hMPV BJ1816 and BJ1887 did not successfully express the proteins (data not shown). Therefore, the 6× his-tag was added to the C terminus of the N genes, and expressed proteins from both recombinant baculoviruses were obtained. The expressed N proteins in Sf21 cells could be identified by strong apple-green fluorescent signals in infected cells using mouse anti-hMPV N protein monoclonal antibody (Fig. 2), whereas there was no apple-green fluorescence observed in cells stained with anti-RSV or polyclonal antibodies against parainfluenza virus 2/3 N protein (Fig. 3), although the signals could be seen in cells infected with the corresponding viruses (data not shown), indicating that there was no cross-reaction among N proteins from hMPV, RSV and parainfluenza virus 2/3. The expressed N proteins (N-His, 44 kDa) were also detected by SDS-PAGE (Fig. 4) and western blotting (Fig. 5). As shown in Fig. 4, N proteins of the expected molecular weight (44 kDa) from both clusters BJ1816 and BJ1887 migrated at the same position but were absent in the lysate from mock-infected Sf21 cells and cells infected with recombinant baculovirus obtained from pFastbac1 without any gene insert.

Immunofluorescent assay for identifying recombinant hMPV N protein a Sf21 cells infected with recombinant baculovirus obtained from pFastbac1 without any gene insert and incubated with mouse anti-hMPV N protein monoclonal antibody as negative control. b Sf21 cells infected with recombinant N BJ1816 -His baculovirus at 72 h post-infection and incubated with mouse anti-hMPV N protein monoclonal antibody. Cells expressing hMPV N protein (in b) show apple-green fluorescence, and cells that do not express hMPV N protein appear red in immunofluorescent microscopy (color figure online)

Immunofluorescent assay for testing cross-reaction among N proteins from hMPV, RSV and parainfluenza virus. Sf21 cells infected with recombinant N BJ1816 -His baculovirus were incubated with: a anti-hMPV N protein monoclonal antibody; b anti-RSV N protein polyclonal antibody; c anti-parainfluenza virus 2/3 N protein polyclonal antibody and showed apple-green fluorescence in a, but not in b and c, indicating that there was no cross-reaction among N proteins of hMPV, RSV and parainfluenza virus (color figure online)

SDS-PAGE analysis of the expression of hMPV N protein Lane M, molecular mass markers (Unstained Protein Molecular Weight Marker, Fermentas, Canada). Lane 1 Sf21 cells infected with recombinant NBJ1816-His baculovirus. Lane 2 N BJ1816 -His protein purified on an Ni column. Lane 3 Sf21 cells infected with recombinant N BJ1887 -His baculovirus. Lane 4 N BJ1887 -His protein purified on an Ni column. Lane 5 Sf21 cells infected with recombinant pfastbac1 baculovirus. Lane 6 mock-infected Sf21 cells (N protein indicated by an arrow). The gel was stained with Coomassie blue

Western blot analysis of recombinant hMPV N proteins. Use of anti-hMPV N polyclonal antibody (a) and anti-his-tag monoclonal antibody (b) to identify expressed recombinant hMPV N proteins. Lane M molecular mass markers (Prestained Protein Marker, NEB, USA). Lane 1 N BJ1816 -His protein purified on an Ni column. Lane 2 N BJ1887 -His protein purified on an Ni column. Lane 3 Sf21 cells infected with recombinant pfastbac1 baculovirus. Lane 4 mock-infected Sf21 cells. Lane 5 lysate from Sf21 cells infected with recombinant N BJ1816 -His baculovirus without purification. Lane 6 lysate from Sf21 cells infected with recombinant N BJ1887 -His baculovirus without purification
It was found that the highest expression level could be achieved at 3 days post-transfection for the first passage. Approximately, 1–2 mg of recombinant protein was obtained from 108 infected Sf21 cells (data not shown). The dialyzed N protein was purified through a Ni–NTA resin column after 20% ammonium sulfate precipitation [19]. Analysis of the eluted fractions by SDS-PAGE and western blot showed that the N-His protein bound efficiently to the column and that the corresponding fractions contained highly purified protein (Figs. 4, 5).
Detection of antibody against recombinant N protein in human sera
In a preliminary experiment, a 44-kDa band was detected with the serum sample from the child with confirmed hMPV infection by western blot (Fig. 6a). The 44-kDa band on the strip transferred with N BJ1887 -His protein was stronger than that with N BJ1816 -His protein, suggesting that this child was infected by a BJ1887-like hMPV (cluster 1). Then, a total of 187 human serum specimens were tested for IgG against hMPV by western blot assay (Fig. 6b shows representative samples of these 187; Table 1) and ELISA (Fig. 7), using purified N-His protein as antigen. The positive rate for antibody was 76% (142/187) by western blot assay and 65% (121/187) by ELISA. It was found that serum specimens with stronger 44-kDa bands in the western blot had higher OD values (OD490nm >1.0) in ELISA, whereas those without bands in the western blot had a much lower OD value (OD490nm <0.2), indicating that, when using this recombinant N protein as antigen, the detection of antibody was consistent by these two methods (Fig. 7).

Western blot analysis of antibody against hMPV in human serum samples using purified N BJ1816 -His protein as antigen. a The hMPV-positive serum sample from the child diagnosed with an hMPV infection. Lane M molecular mass markers (Prestained Protein Marker, Fermentas, USA). Lane 1 Sf21 cells infected with recombinant N BJ1887 -His baculovirus. Lane 2 Sf21 cells infected with recombinant pfastbac1 baculovirus. Lane 3 mock-infected Sf21 cells. b Representative serum samples from 187 serum samples incubated with purified N BJ1816 -His protein (top) and N BJ1887 -His protein (bottom)

Direct-viewing chart of ELISA results for 187 human sera. The bars represent the number of samples in every OD value range, the blue part of the bar represents the number of positive sample, and the red part of the bar represents the number of negative samples in the western blot. The results for antibody detection were consistent between the two methods. The number of positive samples increased with the increase in OD values and showed one cutoff value (about 0.4)
Statistical analysis
The data (Table 1) were analyzed by chi-square test (χ2 test) and consistency checking (Kappa value test). The seropositive rate for each age group by western blot was 41.7% (20/48, <18 years), 84.8% (28/33, 18–30 years), 87.8% (65/74, 31–60 years) and 90.6% (29/32, >60 years). The seropositive rate for the <18 year age group was clearly lower than for the other groups (χ2 = 41.5, P < 0.05). There was no significant difference in the seropositive rate between females and males (χ2 = 0.901, P > 0.05). Although some of the serum samples showed different results, as shown in Table 1 and Fig. 6b, which implied that the immune response to N proteins from these two gene clusters could be different for some individuals, the kappa value was 0.877 (>0.75) when the concordance of results obtained by western blot using N BJ1816 -His protein and N BJ1887 -His protein were compared, revealing that the antigenicity for the N proteins from these two clusters was similar, and therefore, either the N BJ1816 -His protein or N BJ1887 -His protein was sufficient to detect an immune response elicited by hMPV infection. The kappa value was 0.62 (>0.4) when the results from western blot and ELISA using N BJ1816 -His protein only were compared. The result from ELISA was well associated with that obtained by western blot, suggesting a potential alternative antigen for hMPV antibody detection by ELISA.
Discussion
In general, the most conserved gene or gene coding protein is used as a diagnostic target because it can detect a wide variety of related strains [20]. The N protein of hMPV is a major antigen whose amino acid sequence is highly conserved within and between genetic clusters of viruses. It has been shown that there is no cross-reaction between N proteins from hMPV and RSV, although these two viruses can cause similar respiratory illness and co-circulate in the community [14]. Data from this study also support this point of view and exclude cross-reaction among N proteins from hMPV, RSV and parainfluenza virus 2/3 in the family Paramyxoviridae. Several studies had shown the usefulness of recombinant N protein for serological diagnosis of hMPV [11–14]; however, all of them used lysate containing N protein, which is not good enough for future application such as basic research and development of diagnostic kits or vaccines, because it is difficult to guarantee the stability of the protein. In order to improve the expressed protein, a 6× his-tag was added to the C terminus of the gene in a recombinant baculovirus clone. When fused to the C terminus of the hMPV N protein, the 6× his-tag did not affect the expression and yield of the N protein; however, when it was inserted at the N terminus at the very beginning of the study, this protein was not expressed in Sf 21 cells infected with the recombinant baculovirus, as indicated by a lack of signal in a western blot using rabbit anti-hMPV N polyclonal antibody and mouse anti-His monoclonal antibody. This might be because the 6× his-tag at the N terminus changed the structure of the N gene so as to block expression of the protein or because the protein was degraded after expression. In this study, immunofluorescent assay, western blot and ELISA all suggested that the recombinant N-His protein exhibited the epitopes and had the conformation necessary for specific antigen–antibody recognition. In addition, the 6× his-tag insertion at the C terminus made it easier to obtain the purified N protein. This diminishes the difference in results due to using N protein obtained from E. coli or cell lysates containing N protein. Based on this study, it should now be possible to develop an immunoassay for detecting antibodies against hMPV and to use N-His protein for further analysis of the structure and function of the N protein of this virus [19].
To evaluate the antigenicity and practical value of this purified N-His protein, 188 human sera collected from different age groups in Beijing, China, were used for testing antibody against hMPV. By western blot analysis, a high seroprevalence of hMPV antibody could be detected, and the seropositive rate in patients younger than 18 years was clearly lower than that of those older than 18 years. The rate of hMPV exposure in Beijing, China, is similar to that reported by scientists in other countries, including the Netherlands [1], Japan [21] and Canada [13]. In this study, the purified N BJ1887 -His protein was also used as an antigen in ELISA, and the results from western blot and ELISA were consistent, suggesting the potential use of this recombinant protein in the development of an ELISA, which is more convenient for routine laboratory diagnosis and large-scale epidemiological studies.
Although the formation of herringbone- and ring-like subnucleocapsid structures has also been reported for proteins of other paramyxoviruses [19, 22–24], we did not find nucleocapsid-like particles similar in morphology to those described in earlier studies using purified N-His protein—only some aggregation was observed by electron microscopy (data no shown). Efforts to make N proteins that self-assemble and form hMPV virus-like particles are under way.
References
van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD (2001) A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7:719–724. doi:10.1038/89098
Prins JM, Wolthers KC (2004) Human metapneumovirus: a new pathogen in children and adults. Neth J Med 62:177–179
Pelletier G, Dery P, Abed Y, Boivin G (2002) Respiratory tract reinfections by the new human metapneumovirus in an immunocompromised child. Emerg Infect Dis 8:976–978
Cane PA, van den Hoogen BG, Chakrabarti S, Fegan CD, Osterhaus AD (2003) Human metapneumovirus in a haematopoietic stem cell transplant recipient with fatal lower respiratory tract disease. Bone Marrow Transplant 31:309–310. doi:10.1038/sj.bmt.1703849
Ulloa-Gutierrez R, Skippen P, Synnes A, Seear M, Bastien N, Li Y, Forbes JC (2004) Life-threatening human metapneumovirus pneumonia requiring extracorporeal membrane oxygenation in a preterm infant. Pediatrics 114:e517–e519. doi:10.1542/peds.2004-0345
Bastien N, Normand S, Taylor T, Ward D, Peret TC, Boivin G, Anderson LJ, Li Y (2003) Sequence analysis of the N, P, M and F genes of Canadian human metapneumovirus strains. Virus Res 93:51–62
Viazov S, Ratjen F, Scheidhauer R, Fiedler M, Roggendorf M (2003) High prevalence of human metapneumovirus infection in young children and genetic heterogeneity of the viral isolates. J Clin Microbiol 41:3043–3045. doi:10.1128/JCM.41.7.3043-3045.2003
Zhu RN, Qian Y, Deng J, Wang F, Hu AZ, Lu J, Cao L, Yuan Y, Cheng HZ (2003) Human metapneumovirus may associate with acute respiratory infections in hospitalized pediatric patients in Beijing, China. Chin J Pediatr (Zhonghua Er Ke Za Zhi) 41:441–444
Boivin G, Abed Y, Pelletier G, Ruel L, Moisan D, Côté S, Peret TC, Erdman DD, Anderson LJ (2002) Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J Infect Dis 186:1330–1334. doi:10.1086/344319
Cote S, Abed Y, Boivin G (2003) Comparative evaluation of real-time PCR assays for detection of the human metapneumovirus. J Clin Microbiol 41:3631–3635. doi:10.1128/JCM.41.8.3631-3635.2003
Ishiguro N, Ebihara T, Endo R, Ma X, Kawai E, Ishiko H, Kikuta H (2006) Detection of antibodies against human metapneumovirus by western blot using recombinant nucleocapsid and matrix proteins. J Med Virol 78:1091–1095
Hamelin ME, Boivin G (2005) Development and validation of an enzyme-linked immunosorbent assay for humanmetapneumovirus serology based on a recombinant viral protein. Clin Diagn Lab Immunol 12:249–253. doi:10.1128/CDLI.12.2.249-253.2005
Liu L, Bastien N, Sidaway F, Chan E, Li Y (2007) Seroprevalence of human metapneumovirus (hMPV) in the Canadian Province of Saskatchewan Analyzed by a recombinant nucleocapsid protein-based enzyme-linked immunosorbent assay. J Med Virol 79:308–313
Alvarez R, Jones LP, Seal BS, Kapczynski DR, Tripp RA (2004) Serological cross-reactivity of members of the Metapneumovirus genus. Virus Res 105:67–73
Zhu RN, Qian Y (2004) Sequence analysis of the N genes from human metapneumovirus newly identified from children in Beijing. Chin J Microbiol Immunol 2:81–84
Alvarez R, Njenga MK, Scott M, Seal BS (2004) Development of a nucleoprotein-based enzyme-linked immunosorbent assay using a synthetic peptide antigen for detection of avian metapneumovirus antibodies in Turkey sera. Clin Diagn Lab Immunol 11:245–249. doi:10.1128/CDLI.11.2.245-249.2004
Luckow VA, Lee SC, Barry GF, Olins PO (1993) Efficient generation of infectious recombinant baculoviruses by site-specific transposon mediated insertion of foreign genome propagated in Escherichia coli. J Virol 67:4566–4579
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor, New York
Eshaghi M, Tan WS, Ong ST, Yusoff K (2005) Purification and characterization of Nipah virus nucleocapsid protein produced in insect cells. J Clin Microbiol 43:3172–3177. doi:10.1128/JCM.43.7.3172-3177.2005
Samal SK, Pastey MK, McPhillips T, Carmel DK, Mohanty SB (1993) Reliable confirmation of antibodies to bovine respiratory syncytial virus (BRSV) by enzyme-linked immunosorbent assay using BRSV nucleocapsid protein expressed in insect cells. J Clin Microbiol 31:3147–3152
Ebihara T, Endo R, Kikuta H, Ishiguro N, Yoshioka M, Ma X, Kobayashi K (2003) Seroprevalence of human metapneumovirus in Japan. J Med Virol 70:281–283
Kho CL, Tan WS, Tey BT, Yusoff K (2004) NDV nucleocapsid: self assembly and length determination domains. J Gen Virol 84:2163–2168
Bhella D, Ralph A, Murphy LB, Yeo RP (2002) Significant differences in nucleocapsid morphology within the paramyxoviridae. J Gen Virol 83:1831–1839
Juozapaitis M, Coiras M, Staniulis J, Sasnauskas K (2006) Synthesis of the human respiratory syncytial virus nucleoprotein in yeast Saccharomyces cerevisiae.Biologia 3:79–82
Acknowledgments
The authors wish to thank Professor Yan Li from the Department of Medical Microbiology, University of Manitoba, Canada, Influenza and Respiratory Viruses Section, National Microbiology Laboratory, Public Health Agency of Canada, Canada for providing the rabbit anti-hMPV N hyperimmune serum and Sf21 cells. This study was supported by the National Natural Science Foundation of China (30570080; 30872153), Beijing Municipal Natural Science Foundation (7052020) and Beijing outstanding personnel training grant (2006A63) from the Beijing Municipal Committee for Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, L., Qian, Y., Zhu, R. et al. Generation of recombinant nucleocapsid protein of human metapneumovirus in baculovirus for detecting antibodies in the Beijing population. Arch Virol 155, 47–54 (2010). https://doi.org/10.1007/s00705-009-0551-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-009-0551-1