
Abstract
Infection of the CNS with the SARS-CoV-2 can occur via different routes and results in para- or post-infectious manifestations with a variety of neurological symptoms. In patients with neurodegenerative diseases, SARS-CoV-2 is often associated with a higher fatality rate, which is a relevant problem in increasingly older populations. Apart from the direct consequences of an infection in patients with neurodegenerative diseases, indirect consequences of the pandemic such as limited access to care facilities and treatment have negative effects on the course of these chronic disorders. The occurrence of long-lasting neurological symptoms after infection with SARS-CoV-2 indicates a prolonged impact on the CNS. However, while it is known that SARS-CoV-2 affects neuronal populations that are relevant in the pathogenesis of neurodegenerative diseases, it is yet unclear whether an infection with SARS-CoV-2 is sufficient to trigger neurodegeneration. Reflecting on the impact of SARS-CoV-2 on neurodegeneration, we provide a concise overview on the current knowledge of SARS-CoV-2-induced pathology in the CNS and discuss yet open questions in the field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
At the time of writing, almost 500 million people worldwide have been diagnosed with coronavirus disease 2019 (COVID-19), caused by the novel severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) (WHO 2022). Early in the pandemic, it became clear that SARS-CoV-2 infections harbor the risk of developing neurological manifestations (Ababneh et al. 2020; Mao et al. 2020). It has also become evident that SARS-CoV-2 infections have a negative effect on the outcome of patients with neurodegenerative diseases, predominantly studied in common neurodegenerative diseases such as Alzheimer’s disease (AD) or Parkinson’s disease (PD) (Hu et al. 2021, 2022). In PD, an increased mortality rate was associated with SARS-CoV-2 infection, and an increased hospitalization and mortality rate has been shown for patients with AD (McAlpine et al. 2021; Zhang et al. 2020, 2021a). Data on the clinical outcome of SARS-CoV-2 infections on patients with other neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), or Huntington Disease (HD) are yet limited and will require further studies in the course of the pandemic (De Marchi et al. 2021; Musson et al. 2022). Also, chronic neurological diseases such as multiple sclerosis with increased disability and a progressive course of disease have been associated with higher mortality rates (Prosperini et al. 2022; Barzegar et al. 2021).
So far, different mechanisms have been considered as a cause of neurological symptoms or acceleration of existing neurological conditions during SARS-CoV-2 infection. These include direct effects of the virus on the central nervous system (CNS) by, e.g., entering the brain via nasal or oral routes and infection of neuronal populations (Meinhardt et al. 2021) and para- or post-infectious effects such as triggering inflammation and auto-immune reactions (Zubair et al. 2020; Franke et al. 2021). Those effects of SARS-CoV-2 on the CNS have potential implications for the development of long-term neurological conditions including neurodegeneration. In addition, systemic effects during severe sepsis can trigger neurological symptoms (Heneka et al. 2020; Widmann and Heneka 2014), and depend on the outcome and management of the acute disease.
In this review article, we will summarize recent evidence of effects of SARS-CoV-2 infection on the CNS and its implications for neurodegenerative diseases. We identified the existing evidence through author knowledge and PubMed searches from database inception, to March, 2022 using the search terms: “neurofilament”, “neurodegeneration”, “neurodegenerative diseases”, “Parkinson disease”, “Alzheimer disease”, “amyotrophic lateral sclerosis”, “dementia”, “multiple sclerosis”, “Covid-19”, and “SARS-CoV-2”.
Nervous system involvement in SARS-CoV-2-infection
When COVID-19 emerged, it became evident that the involved pathogen, the novel SARS-CoV-2 variant, was more transmittable between humans than its predecessor SARS-CoV-1 (Liu et al. 2020b). Similar to its predecessor, SARS-CoV-2 uses the same densely glycosylated spike (S) protein on the viral surface to bind to the peptidase domain of the angiotensin-converting enzyme 2 receptor (ACE2) on mammalian host cells to enter cells yet with much higher affinity (Wrapp et al. 2020). ACE2 is an extracellular enzyme with a transmembrane domain located toward the cell surface. Upon protease activation, soluble ACE2 is released into the extracellular space, where it physiologically processes angiotensin peptides as part of the angiotensin–renin pathway, while the transmembrane domain of ACE2 is internalized into the cell. During the internalization process, SARS-CoV-2 bound to ACE2 is either taken up by clathrin-coated vesicles or directly through involvement of the ACE2-specific transmembrane protease serine subtype 2 (TMPRSS2) (Wu et al. 2021). ACE2 is predominantly expressed in airway epithelia and lung parenchyma, kidney cells, small intestine, and vascular endothelia throughout the body, but also widely throughout the CNS (Deinhardt-Emmer et al. 2021). Predilection sites of ACE2 in the brain are the posterior cingulate gyrus, motor cortex, olfactory bulb, middle temporal gyrus, substantia nigra, and the ventricles, where it is expressed in neurons, astrocytes, and oligodendrocytes (Xia and Lazartigues 2008; Chen et al. 2020).
Loss of smell and taste were among the first CNS-related symptoms associated with SARS-CoV-2 infection, providing the early support for the hypothesis of direct entry of SARS-CoV-2 into the brain via the olfactory and oral mucosa. Although loss of smell is a common symptom in COVID-19 patients, its manifestation likely depends on the genetic variability of the virus and its host such as differences in ACE2 and TMPRSS2 expression (Li et al. 2020; Korber et al. 2020; Shang et al. 2020; von Bartheld et al. 2020; Strafella et al. 2020; Williams et al. 2020). Studies with SARS-CoV-infected mice showed that virus antigen is found in the olfactory bulb and subsequently in connected brain regions such as the piriform cortex, basal ganglia, midbrain, and cardiorespiratory region of the medulla (Netland et al. 2008). The detection of SARS-CoV-2 in human brain tissue has been challenging and likely depends on the severity of the CNS involvement. Whereas no evidence of SARS-CoV-2 was found in the brain of 19 infected patients without neurological symptoms (Hirschbuhl et al. 2021), a post-mortem study of 33 SARS-CoV-2 patients with neurological symptoms found viral RNA in the nasal mucosa and to a lesser extend in the ophthalmic and oral mucosa as well as in the medulla oblongata (Meinhardt et al. 2021). This study suggests that infection of the CNS by SARS-CoV-2 in patients with neurological symptoms is mediated by transsynaptic transmission across peripheral olfactory neurons to connected brain areas. Although this study did not detect SARS-CoV-2 in the carotid blood vessels and thus argued against a systemic infection via the blood stream, this route of infection cannot be completely ruled out. In a more detailed study of the olfactory mucosa, the virus was found in sustentacular and stem cells of the mucosa while absent from olfactory receptor neurons (ORN) (Butowt and von Bartheld 2020).
Alternative routes of CNS infection have been described across the blood–brain barrier (BBB), ACE2 expressing vascular endothelial cells, or indirect trafficking of the virus via blood leukocytes under increased permeability of the BBB (Zubair et al. 2020; Gomes et al. 2021). By infecting inflammatory cells such as monocytes, macrophages, dendritic cells, and lymphocytes, the virus is able to initiate a cascade of cytokine release and an overwhelming activation of leucocytes which is responsible for subsequent cellular damage. Alternative explanations for SARS-CoV-2 CNS pathogenicity come from its association with auto-immune diseases (Tang et al. 2021). Its complex transcriptome, its ability to mimic human proteins, its ability to promote the generation of autoantibodies, and activate cytokine responses trigger auto-immune responses (Kim et al. 2020; Kanduc 2021; Zhou et al. 2020c). Neuroimmune manifestations such as Guillain–Barré syndrome (GBS), myasthenia gravis, acute disseminated encephalomyelitis (ADEM), or encephalitis suggest an autoantibody-mediated inflammatory response in certain neuronal populations (Toscano et al. 2020; Parsons et al. 2020; Zhou et al. 2020b; Muhammed et al. 2021; Restivo et al. 2020; Foresti et al. 2021). In blood and CSF of SARS-CoV-2-infected patients with neurological symptoms, known autoantibodies against the NMDA-receptor or intracellular Yo-antigen were found, but also antibody binding on yet undetermined antigen epitopes on vessel endothelium, astrocytic proteins, neuropil of basal ganglia, hippocampus, or olfactory bulb from such samples was discovered (Watson et al. 2021; Franke et al. 2021; Mulder et al. 2021). This implies immunosuppressive therapy or plasmapheresis as a possible treatment for autoantibody-associated neurological symptoms.
SARS-CoV-2-triggered neurological symptoms and complications
Neurological symptoms have been described during and post-infection with SARS-CoV-2 (Rifino et al. 2021; Ellul et al. 2020). The clinical presentation of neurological and encephalopathic findings ranged from mild headache and confusion over fluctuating levels of consciousness and extrapyramidal movement disorders (e.g., myoclonus) to seizures and coma (Garg et al. 2021; Romero-Sanchez et al. 2020). Although the number of neurological complications might be lower in SARS-CoV-2 compared to other viruses, the scale of the pandemic renders the overall number of neurological complications and diseases a relevant socioeconomic burden (Ellul et al. 2020). As direct evidence of viral DNA cannot be measured in the CNS of living patients, cerebrospinal fluid is used as an alternative source. However, only two case reports reported positive results for SARS-CoV-2 DNA in the CSF of COVID-19 patients with associated encephalitis, while larger retrospective studies of COVID-19 patients with neurological diseases had negative CSF measurements (Moriguchi et al. 2020; Zhou et al. 2020a; Destras et al. 2020; Neumann et al. 2020; Bellon et al. 2021). Nevertheless, several case studies confirmed neurological diseases associated with SARS-CoV-2 infection. Mild encephalopathies with transient symptoms such as altered consciousness (19.1% of 841 patients) or bradypsychia and disorientation (10.1% of 841 patients) were found to occur rather commonly often accompanied by findings of unspecific T2/FLAIR hyperintensity (35% of 20 patients) and ischemic infarcts (31% of 108 patients) in neuroimaging (Romero-Sanchez et al. 2020; Mahammedi et al. 2020; Radmanesh et al. 2020). Severe COVID-19-associated encephalopathies including ADEM or acute necrotizing encephalopathies and poly(radiculo)neuropathies as GBS or other acute neuropathies (multifocal demyelinating or small fiber polyneuropathy) have also been described in several case reports with an estimated prevalence of 0.1–1% in western countries within 6 weeks of confirmed infection (Mahapure et al. 2021; Parsons et al. 2020; Shahali et al. 2021; Toscano et al. 2020; Camdessanche et al. 2020; Hayley and Sun 2021; Dixon et al. 2020; Poyiadji et al. 2020; Oaklander et al. 2022). For some entities, a causative relationship to prior SARS-CoV-2 infection is less clear. For example, a large prospective observational study between January and May 2020 could not confirm a higher risk for GBS after SARS-CoV-2 infection (Luijten et al. 2021) and another study of 145.721 cases with COVID-19 showed that GBS had a low pooled prevalence (0.28% within the study cohort) compared to other neurological manifestations (Misra et al. 2021). Among the most common neurological complications in hospitalized patients with COVID-19 are cerebrovascular diseases which are likely associated with a pro-inflammatory hypercoagulable state in severe infection with elevated C-reactive protein, D-dimer, and ferritin (Lodigiani et al. 2020; Benussi et al. 2020). Patients presented with ischemic and hemorrhagic stroke or venous thrombosis. In a recent meta-analysis of 145,634 COVID-19 patients, of which 89% were hospitalized, every third patient presented with a neurological manifestation (Misra et al. 2021). A cross-sectional surveillance study in neurology, stroke, psychiatry, and intensive-care units in the UK in April 2020 included 125 patients of which 77 (62%) had a cerebrovascular event (74% ischemic, 12% intracerebral hemorrhage, and 1% vasculitis), while altered mental status was the second most common presentation (Varatharaj et al. 2020). In 267 cases that were followed-up during that time COVID-19 associated stroke was mostly observed in younger adults, delirium was common in patients over 60 years and encephalopathy mostly in patients under 60 years treated on intensive-care units (Ross Russell et al. 2021). Although less frequent, infections with SARS-CoV-2 have also been reported to result in movement disorders (Salari et al. 2021; Hull et al. 2021).
Implications for patients with neurodegenerative disorders
Populations at risk for a deleterious outcome of COVID-19 have been identified early in the pandemic and encompass patients with higher age, male sex, obesity, diabetes, and other comorbidities (Williamson et al. 2020). Patients with neurodegenerative disease are at particular risk, being mostly of higher age and presenting with many relevant comorbidities (Kitani-Morii et al. 2021). Several studies have shown that a pre-existing neurodegenerative disease itself worsens SARS-CoV-2 infection outcomes. Data derived from meta-analyses and the UK Biobank have shown that dementia is an age-independent risk factor for a deleterious outcome of COVID-19 with increased disease severity and higher likelihood of death compared to non-demented control patients (Tahira et al. 2021; Liu et al. 2020a). This effect has been confirmed for the subgroup of patients with AD, while vascular dementia was not associated with a higher risk (Zhang et al. 2021b). The clinical presentation of COVID-19 infection in patients with concomitant dementia is often atypical with apathy and confusion or hyperactive delirium as leading symptoms, while cough, fever, or dyspnea are less frequent symptoms (Bianchetti et al. 2020; Harb et al. 2021; Mendes et al. 2021; Rebora et al. 2021; Hariyanto et al. 2021). In addition, delirium is an independent factor for worsening the outcome of SARS-CoV-2 including increased fatality (Rozzini et al. 2020; Garcez et al. 2020; Mendes et al. 2021). In most retrospective analyses, a higher case fatality rate has been reported in patients with PD. Particularly, PD patients with longer disease duration and higher age show a high mortality rate of 21–40% during SARS-CoV-2 infections in different cohorts in Italy, the US, and Germany and independent of age, sex and ethnic background (Antonini et al. 2020; Zhang et al. 2020; Scherbaum et al. 2021). On the other hand, some studies with smaller sample sizes recruiting patients in tertiary referral centers in Europe and Italy did not find an increased mortality rate in PD nor dementia compared to demographically matched controls (Huber et al. 2021; Fasano et al. 2020).
Data on other neurodegenerative diseases are less extensive. A study using the US veteran database and 699 patients with amyotrophic lateral sclerosis (ALS) found an increased risk of COVID-19 related death in this cohort (Galea et al. 2021). In a Scottish cohort of 1062 patients with motor neuron disease, 77.7% of whom were diagnosed with ALS, all-cause mortality remained unaffected in 2020, though specific mortality rates of individuals diagnosed with COVID-19 are not reported (Glasmacher et al. 2021). Interestingly, C9orf72 repeat expansions of intermediate length, a mutation often found in cases of familial ALS and frontotemporal dementia (FTD), have been associated with severe COVID-19 requiring mechanical ventilation (Zanella et al. 2021). Multiple sclerosis itself has not been associated with a severe course of disease, whereas increased disability and a progressive course of the disease, which is mainly mediated by neurodegenerative processes, have (Prosperini et al. 2022; Barzegar et al. 2021). Future prospective studies including all healthcare services may shed light on the actual risk of patients with neurodegenerative disorders.
Besides the direct effect of an infection with SARS-CoV-2 on patients with neurodegenerative diseases, the impact of the global COVID-19 pandemic on these patients due to disruptions in the medical care is equally alarming. Since onset of the global pandemic in late 2019, symptoms such as anxiety, depression, and sleep disturbances have been found to have increased in the general public (Thygesen et al. 2021; Bauerle et al. 2020). This is in line with cross-sectional data from more than 5000 patients with PD that revealed worsening of motor (43%) and especially non-motor (52%) symptoms since pandemic onset (Brown et al. 2020). Home confinement and other disease control measures aiming at reduction of contacts are likely to contribute to a diminished physical activity in PD patients (Leavy et al. 2021) and foster the discontinuation of medical treatments, such as multimodal complex treatment or levodopa/carbidopa intestinal gel (LCIG) set-ups (Richter et al. 2021; Wolff et al. 2022). Disruptions were furthermore observed to impact clinical trials, due to difficulties in recruitment, initiation and monitoring (Lorusso et al. 2020) which will impact the development of future therapeutic options. Although adaptions such as implementation of remote monitoring and remote study endpoints have been made, they need to be expanded in the future (Snider and Holtzman 2021).
Potential long-term consequences of SARS-CoV-2 infections
After acute infection with SARS-CoV-2, some of the associated symptoms can persist or additional symptoms can emerge. The British National Institute for Health and Care Excellence (NICE) distinguishes on one hand ongoing symptomatic COVID-19, defined as persistence of COVID-19-related signs and symptoms up to 12 weeks after infection, from the post-COVID-19 syndrome, defined as the persistence or occurrence of COVID-19-related symptoms 12 weeks after infection (Fig. 1) (NICE 2022). The term “long-COVID” refers to the entirety of symptoms after an acute SARS-CoV-2 infection independent of time and duration and can be considered a disability under the Americans with Disabilities Act (ADA) since July 2021 (Centers for Disease and Prevention 2021; HHS.gov 2021). Patients suffering from post-COVID-19 syndrome may present with a volatile cluster of symptoms in any system of the body. These may include typical COVID-19-related symptoms such as loss of smell or dyspnea, but also include neurological symptoms, such as cognitive impairment, dizziness, and delirium (NICE 2022). Longitudinal assessment of 9751 patients with COVID-19 found the persistence of COVID-19-related symptoms in 72.5% of the cases after discharge (follow-up range from 29 to 234 days), most prominently shortness of breath or dyspnea (36.0%), and fatigue or exhaustion (40.0%) (Nasserie et al. 2021; Lara et al. 2020). Particularly, the persistence of fatigue or muscle weakness correlated with the need of supplemental oxygen or the need of invasive and non-invasive breathing assistance in hospitalized COVID-19 patients 6 months after discharge (Huang et al. 2021). Both, the pandemic itself, and a previous infection with SARS-CoV-2 have been linked to an increase in distress and depression in the general public (Ramiz et al. 2021; Nasserie et al. 2021). In particular, patients with pre-existing neurodegenerative or chronic neurological diseases are at special risk for this development. Patients with dementia, AD, PD, and multiple sclerosis have been found to suffer from worsening of pre-existing symptoms and de-novo development of neuropsychiatric symptoms, e.g., anxiety and cognitive decline (Lara et al. 2020; Boutoleau-Bretonniere et al. 2020; Wei et al. 2022; Salari et al. 2020; Shalash et al. 2020; Wolff et al. 2022; Haji Akhoundi et al. 2020). This long-lasting impact of an infection with SARS-CoV-2 has resulted in the need for an implementation of a medical care structure for these patients (Gemelli Against 2020).

Terms used to describe persisting symptoms after SARS-CoV-2 infection. NICE, British National Institute for Health and Care Excellence; ADA, Americans with Disabilities Act
The association of neuropsychiatric symptoms with SARS-CoV-2 infection raises the question about the potential long-term impact of the virus on the CNS, its molecular basis, and the potential risk of neuronal damage associated with subsequent development of neurodegenerative diseases. During SARS-CoV infection, there is evidence of scattered degeneration of neurons as a sign of neuronal hypoxia/ischemia (Gu et al. 2005). Sepsis itself has long-term consequences on the brain often associated with delirium and even cognitive decline later in life. Several mechanisms have been discussed to cause neuronal damage during sepsis. First, decreased blood flow caused by reduction in peripheral vascular tone and diminished cardiac output alters brain circulation. Altered microcirculation of the brain, possibly also through an impaired autoregulation caused by increased CO2 levels, leads to detachment of pericytes from the basal lamina and increased permeability of the BBB (Nishioku et al. 2009; Barichello et al. 2021). Sepsis triggers the peripheral innate immune system with an increased secretion of TNF-alpha and interleukins causing endothelial cell damage, disintegration of intracellular junctions, and increased permeability of the BBB. Invasion of T-lymphocytes and macrophages via the BBB activates microglia and astrocytes leading to cerebral cytokine, chemokine, and nitric oxide release, which causes neuronal dysfunction and ultimately cell death (Heneka et al. 2020; Widmann and Heneka 2014). A well-established indicator for neuronal damage is the increase of blood or CSF levels of neurofilament light chain (NfL), which is a structural protein and part of the cytoskeleton of long axons. An increase in neurofilament levels has been related to unspecific neuronal damage that can be caused by neuroinflammation, neuronal ischemia, or neurodegeneration (Khalil et al. 2018). Several studies assessed neurofilament levels in SARS-CoV-2 patients. CSF and blood NfL levels were elevated in patients with severe disease and concomitant neurological symptoms (Virhammar et al. 2021; Paterson et al. 2021). Another study evaluated blood NfL levels of 47 SARS-CoV-2 patients at admittance to hospital and showed that a higher baseline NfL level is a negative predictor of survival (Aamodt et al. 2021). Blood NfL was increased in the acute phase of 48 severe and 28 moderate SARS-CoV-2 cases, while after a 6-month follow-up NfL was normalized (Kanberg et al. 2021). To predict the extent of neuronal damage in a cohort of 142 hospitalized patients with COVID-19, Prudencio et al. measured serum NfL at time of hospitalization and at multiple follow-ups (Prudencio et al. 2021). Higher baseline NfL levels indicated worse clinical outcome, while over time NfL levels stayed constant or fluctuated, without a clear correlation to clinical symptoms. Although neurofilaments are an indicator of neuronal damage, it is not yet clear if their elevation indicates a risk for long-term neurodegeneration. To further support neuronal loss, diminished gray matter within the limbic cortical areas with direct neuronal connectivity to the primary olfactory system has been recently described in longitudinal brain imaging after COVID-19 infection (Douaud et al. 2021).
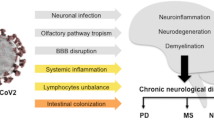
Further evidence of a potential risk for the development of neurodegenerative diseases comes from case reports describing patients with akinetic-rigid parkinsonism following infection with SARS-CoV-2. In addition to improvement of symptoms either under Levodopa/Carbidopa-treatment or spontaneously, all patients displayed abnormal imaging findings, such as decreased dopamine transporter density in dopamine transporter scan or decreased dopamine uptake in 18F-DOPA PET (Cohen et al. 2020; Faber et al. 2020; Mendez-Guerrero et al. 2020). It remains uncertain, if these patients developed the clinical findings de-novo or if a subclinical PD emerged after worsening through severe COVID-19. Causal links of viral infections to neurodegenerative diseases have been drawn since the influenza pandemic in 1918, which is assumed to have caused numerous cases of postencephalitic parkinsonism (Kim et al. 2015; Hoffman and Vilensky 2017; Bigman and Bobrin 2018). This theory was further fueled by the fact that the substantia nigra represents a potential target for neurovirulent influenza A viruses (Takahashi et al. 1995). This is also true for coronaviruses, which have been shown to accumulate in the nucleus subthalamicus and the substantia nigra after infection in mice (Fishman et al. 1985). Furthermore, elevated titers of antibodies directed against coronaviruses have been found in patients with PD (Fazzini et al. 1992). ACE2 has not only been found to be expressed by midbrain dopaminergic neurons, but dopaminergic neurons were also permissive for an infection with SARS-CoV-2 in-vitro (Yang et al. 2020). Further evidence for the involvement of the midbrain during COVID-19 was added by the fact that post-mortem analysis of patients with COVID-19 revealed activated microglia and cytotoxic T-lymphocytes in the brainstem and cerebellum (Matschke et al. 2020). By infiltration of viruses to the CNS, neuroinflammation can be triggered, resulting in a disruption of the BBB, microglia activation, and clustering of microglia around (dopaminergic) neurons, as well as pro-inflammatory cytokine release. This is thought to result in a vicious circle of self-driven neuroinflammation, eventually resulting in neurodegeneration (Limphaibool et al. 2019). In PD, there is strong evidence suggesting alpha-synuclein pathology propagates from the olfactory bulb and/or the gut to the brain, which is associated with early non-motor symptoms such as olfactory disfunction and obstipation (Braak et al. 2003; Haehner et al. 2011). Olfactory dysfunction, in particular loss of smell, is also a common symptom of COVID-19. ACE2 and TMPRSS2 have been shown to be expressed in human olfactory mucosa and therefore pose a possible entry site of SARS-CoV-2 to the brain, which is shared by alpha-synuclein pathology (Fig. 2; Brann et al. 2020). Additionally, viral infections were found to increase alpha-synuclein expression levels in rodents and to induce the formation of cytotoxic alpha-synuclein aggregates (Beatman et al. 2015; Bantle et al. 2019). Whether and in what way these findings might contribute to the development of PD is unknown. Furthermore, SARS-CoV-2 has been shown to also influence other proteins involved in neurodegenerative diseases such as AD: the expression of SARS-CoV-2 spike protein S has been linked to an increased spreading of Tau and cytosolic prions in-vitro and further prompted the aggregation of these proteins (Liu et al. 2021). A shift in localization of Tau from axons to the soma and hyperphosphorylation of Tau, both hallmarks of early stages of tauopathy, have been observed in SARS-CoV-2-infected neurons, resulting in cell death in 3D human brain organoids (Ramani et al. 2020). In Amyotrophic lateral sclerosis (ALS), the main pathological protein found aggregated in the cytoplasm of neurons leading to progressive degeneration of motor neurons is TAR DNA-binding protein 43 (TDP-43). In 10% of ALS cases, a known gene mutation underlies the disease, while in the majority of sporadic ALS patients, a multi-hit hypothesis of internal and external factors including environmental risk factors has been considered (Brown and Al-Chalabi 2017). A viral infection of specific neuronal populations such as motor neurons might lower the threshold for developing TDP-43 pathology as proposed in the multistep hypothesis for ALS (Chio et al. 2018). In summary, there is growing evidence of COVID-19 associated neuropathological effects and SARS-CoV-2 may be impacting many pathways involved in the pathogenesis of neurodegenerative diseases. Whether this is sufficient to induce neurodegeneration and whether the human nervous system is able to counteract and regenerate is subject of ongoing research. Longitudinal data will be needed to evaluate the relevance of this impact on patients diagnosed with COVID-19.

SARS-CoV-2 and neurodegenerative diseases. a Clinical presentation: SARS-CoV-2 infection leads to neurological symptoms and disorders (e.g., loss of smell, altered mental status, and stroke), severe sepsis, or worsening of neurodegenerative diseases. Whether SARS-CoV-2 induces neurodegenerative disorders is unknown. b–d Cellular and molecular mechanisms. b SARS-CoV-2 enters the brain via the nasal and oral mucosa and affects multiple brain regions, also those involved in neurodegenerative disorders. Systemic inflammation leads to dysfunction of the BBB and neuroinflammation. c Activation of microglia, astrocytes, and pro-inflammatory cytokines in the brain may trigger neuronal damage and release of NfL. d Neuronal damage by SARS-CoV-2 could be a potential risk for the development of neurodegeneration associated with proteinopathy, such as PD, AD and ALS. PD Parkinson’s disease, AD Alzheimer’s Disease, ALS amyotrophic lateral sclerosis, NfL neurofilament light chain, Tdp-43 TAR DNA-binding protein 43, a-Syn alpha-synuclein, Tau Protein Tau (Created with https://biorender.com/)
Open questions
This short review summarizes molecular and clinical evidence of CNS affection by the SARS-CoV-2 virus with particular reference to neurodegenerative disorders. Many questions, however, prevail. Although it is known that COVID-19 can cause neuronal damage as indicated by increased neurofilament levels, it is unresolved whether the CNS is able to recover completely or only partially from this damage and what the delay of such a recovery might be. Even less clear is whether such damage to the CNS triggers neurodegenerative diseases and with which latency the (clinical) onset of such diseases might be expected. Which genetic predispositions or environmental factors could modulate these outcomes? Are all viral strains/variants of concern equally dangerous in this regard? We also do not know whether long-COVID or post-COVID-19 syndrome predisposes to such potential long-term consequences. We know that vaccination against SARS-CoV-2 is a powerful means to prevent a severe course of disease in the general public (Thompson et al. 2021; Lin et al. 2022; Rosenberg et al. 2022; Hippisley-Cox et al. 2021). Data on the safety and effectiveness of the available vaccines in the vulnerable population of patients with neurodegenerative diseases are lacking and urgently needed (Shi et al. 2021). It is also not clear to date whether vaccination also prevents potential neurodegeneration as sequelae of COVID-19 and what the role of different vaccines may be. Only longitudinal studies with large cohorts and appropriate controls will be able to solve these questions in the future (Horn et al. 2021).
References
Aamodt AH, Hogestol EA, Popperud TH, Holter JC, Dyrhol-Riise AM, Tonby K, Stiksrud B, Quist-Paulsen E, Berge T, Barratt-Due A, Aukrust P, Heggelund L, Blennow K, Zetterberg H, Harbo HF (2021) Blood neurofilament light concentration at admittance: a potential prognostic marker in COVID-19. J Neurol. https://doi.org/10.1007/s00415-021-10517-6
Ababneh NA, Scaber J, Flynn R, Douglas A, Barbagallo P, Candalija A, Turner MR, Sims D, Dafinca R, Cowley SA, Talbot K (2020) Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum Mol Genet 29(13):2200–2217. https://doi.org/10.1093/hmg/ddaa106
Antonini A, Leta V, Teo J, Chaudhuri KR (2020) Outcome of Parkinson’s disease patients affected by COVID-19. Mov Disord 35(6):905–908. https://doi.org/10.1002/mds.28104
Bantle CM, Phillips AT, Smeyne RJ, Rocha SM, Olson KE, Tjalkens RB (2019) Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis 5:20. https://doi.org/10.1038/s41531-019-0090-8
Barichello T, Generoso JS, Collodel A, Petronilho F, Dal-Pizzol F (2021) The blood-brain barrier dysfunction in sepsis. Tissue Barriers 9(1):1840912. https://doi.org/10.1080/21688370.2020.1840912
Barzegar M, Mirmosayyeb O, Gajarzadeh M, Afshari-Safavi A, Nehzat N, Vaheb S, Shaygannejad V, Maghzi AH (2021) COVID-19 among patients with multiple sclerosis: a systematic review. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000001001
Bauerle A, Teufel M, Musche V, Weismuller B, Kohler H, Hetkamp M, Dorrie N, Schweda A, Skoda EM (2020) Increased generalized anxiety, depression and distress during the COVID-19 pandemic: a cross-sectional study in Germany. J Public Health (oxf) 42(4):672–678. https://doi.org/10.1093/pubmed/fdaa106
Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, Beckham JD (2015) Alpha-Synuclein expression restricts RNA viral infections in the brain. J Virol 90(6):2767–2782. https://doi.org/10.1128/JVI.02949-15
Bellon M, Schweblin C, Lambeng N, Cherpillod P, Vazquez J, Lalive PH, Schibler M, Deffert C (2021) Cerebrospinal fluid features in severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (RT-PCR) positive patients. Clin Infect Dis 73(9):e3102–e3105. https://doi.org/10.1093/cid/ciaa1165
Benussi A, Pilotto A, Premi E, Libri I, Giunta M, Agosti C, Alberici A, Baldelli E, Benini M, Bonacina S, Brambilla L, Caratozzolo S, Cortinovis M, Costa A, Cotti Piccinelli S, Cottini E, Cristillo V, Delrio I, Filosto M, Gamba M, Gazzina S, Gilberti N, Gipponi S, Imarisio A, Invernizzi P, Leggio U, Leonardi M, Liberini P, Locatelli M, Masciocchi S, Poli L, Rao R, Risi B, Rozzini L, Scalvini A, Schiano di Cola F, Spezi R, Vergani V, Volonghi I, Zoppi N, Borroni B, Magoni M, Pezzini A, Padovani A (2020) Clinical characteristics and outcomes of inpatients with neurologic disease and COVID-19 in Brescia, Lombardy, Italy. Neurology 95(7):e910–e920. https://doi.org/10.1212/WNL.0000000000009848
Bianchetti A, Rozzini R, Guerini F, Boffelli S, Ranieri P, Minelli G, Bianchetti L, Trabucchi M (2020) Clinical presentation of COVID19 in dementia patients. J Nutr Health Aging 24(6):560–562. https://doi.org/10.1007/s12603-020-1389-1
Bigman DY, Bobrin BD (2018) Von Economo’s disease and postencephalitic parkinsonism responsive to carbidopa and levodopa. Neuropsychiatr Dis Treat 14:927–931. https://doi.org/10.2147/NDT.S153313
Boutoleau-Bretonniere C, Pouclet-Courtemanche H, Gillet A, Bernard A, Deruet AL, Gouraud I, Mazoue A, Lamy E, Rocher L, Kapogiannis D, El Haj M (2020) The effects of confinement on neuropsychiatric symptoms in Alzheimer’s disease during the COVID-19 crisis. J Alzheimers Dis 76(1):41–47. https://doi.org/10.3233/JAD-200604
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, Chance R, Macaulay IC, Chou HJ, Fletcher RB, Das D, Street K, de Bezieux HR, Choi YG, Risso D, Dudoit S, Purdom E, Mill J, Hachem RA, Matsunami H, Logan DW, Goldstein BJ, Grubb MS, Ngai J, Datta SR (2020) Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. https://doi.org/10.1126/sciadv.abc5801
Brown EG, Chahine LM, Goldman SM, Korell M, Mann E, Kinel DR, Arnedo V, Marek KL, Tanner CM (2020) The effect of the COVID-19 pandemic on people with Parkinson’s disease. J Parkinsons Dis 10(4):1365–1377. https://doi.org/10.3233/JPD-202249
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377(2):162–172. https://doi.org/10.1056/NEJMra1603471
Butowt R, von Bartheld CS (2020) Anosmia in COVID-19: underlying mechanisms and assessment of an olfactory route to brain infection. Neuroscientist. https://doi.org/10.1177/1073858420956905
Camdessanche JP, Morel J, Pozzetto B, Paul S, Tholance Y, Botelho-Nevers E (2020) COVID-19 may induce Guillain-Barre syndrome. Rev Neurol (paris) 176(6):516–518. https://doi.org/10.1016/j.neurol.2020.04.003
Chen R, Wang K, Yu J, Howard D, French L, Chen Z, Wen C, Xu Z (2020) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front Neurol 11:573095. https://doi.org/10.3389/fneur.2020.573095
Chio A, Mazzini L, D’Alfonso S, Corrado L, Canosa A, Moglia C, Manera U, Bersano E, Brunetti M, Barberis M, Veldink JH, van den Berg LH, Pearce N, Sproviero W, McLaughlin R, Vajda A, Hardiman O, Rooney J, Mora G, Calvo A, Al-Chalabi A (2018) The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology 91(7):e635–e642. https://doi.org/10.1212/WNL.0000000000005996
Cohen ME, Eichel R, Steiner-Birmanns B, Janah A, Ioshpa M, Bar-Shalom R, Paul JJ, Gaber H, Skrahina V, Bornstein NM, Yahalom G (2020) A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol 19(10):804–805. https://doi.org/10.1016/S1474-4422(20)30305-7
COVID-19 rapid guidline: managing the long-term effects of COVID-19 (2021). Accessed 13 Dec 2021
De Marchi F, Gallo C, Sarnelli MF, De Marchi I, Saraceno M, Cantello R, Mazzini L (2021) Accelerated early progression of amyotrophic lateral sclerosis over the COVID-19 pandemic. Brain Sci. https://doi.org/10.3390/brainsci11101291
Deinhardt-Emmer S, Wittschieber D, Sanft J, Kleemann S, Elschner S, Haupt KF, Vau V, Haring C, Rodel J, Henke A, Ehrhardt C, Bauer M, Philipp M, Gassler N, Nietzsche S, Loffler B, Mall G (2021) Early postmortem mapping of SARS-CoV-2 RNA in patients with COVID-19 and the correlation with tissue damage. Elife. https://doi.org/10.7554/eLife.60361
Destras G, Bal A, Escuret V, Morfin F, Lina B, Josset L, Group CO-DHS (2020) Systematic SARS-CoV-2 screening in cerebrospinal fluid during the COVID-19 pandemic. Lancet Microbe 1(4):e149. https://doi.org/10.1016/S2666-5247(20)30066-5
Dixon L, Varley J, Gontsarova A, Mallon D, Tona F, Muir D, Luqmani A, Jenkins IH, Nicholas R, Jones B, Everitt A (2020) COVID-19-related acute necrotizing encephalopathy with brain stem involvement in a patient with aplastic anemia. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000000789
Douaud G, Lee S, Alfaro-Almagro F, Arthofer C, Wang C, McCarthy P, Lange F, Andersson JLR, Griffanti L, Duff E, Jbabdi S, Taschler B, Winkler AM, Nichols TE, Collins R, Matthews PM, Allen N, Miller KL, Smith SM (2021) Brain imaging before and after COVID-19 in UK Biobank. medRxiv. https://doi.org/10.1101/2021.06.11.21258690
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S, Sejvar J, Solomon T (2020) Neurological associations of COVID-19. Lancet Neurol 19(9):767–783. https://doi.org/10.1016/S1474-4422(20)30221-0
Faber I, Brandao PRP, Menegatti F, de Carvalho Bispo DD, Maluf FB, Cardoso F (2020) Coronavirus disease 2019 and Parkinsonism: a non-post-encephalitic Case. Mov Disord 35(10):1721–1722. https://doi.org/10.1002/mds.28277
Fasano A, Cereda E, Barichella M, Cassani E, Ferri V, Zecchinelli AL, Pezzoli G (2020) COVID-19 in Parkinson’s disease patients living in Lombardy, Italy. Mov Disord 35(7):1089–1093. https://doi.org/10.1002/mds.28176
Fazzini E, Fleming J, Fahn S (1992) Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov Disord 7(2):153–158. https://doi.org/10.1002/mds.870070210
Fishman PS, Gass JS, Swoveland PT, Lavi E, Highkin MK, Weiss SR (1985) Infection of the basal ganglia by a murine coronavirus. Science 229(4716):877–879. https://doi.org/10.1126/science.2992088
Foresti C, Servalli MC, Frigeni B, Rifino N, Storti B, Gritti P, Fabretti F, Grazioli L, Sessa M (2021) COVID-19 provoking Guillain-Barre syndrome: The Bergamo case series. Eur J Neurol 28(10):e84–e85. https://doi.org/10.1111/ene.14549
Franke C, Ferse C, Kreye J, Reincke SM, Sanchez-Sendin E, Rocco A, Steinbrenner M, Angermair S, Treskatsch S, Zickler D, Eckardt KU, Dersch R, Hosp J, Audebert HJ, Endres M, Ploner JC, Pruss H (2021) High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. Brain Behav Immun 93:415–419. https://doi.org/10.1016/j.bbi.2020.12.022
Galea MD, Galea VP, Eberhart AC, Patwa HS, Howard I, Fournier CN, Bedlack RS (2021) Infection rate, mortality and characteristics of veterans with amyotrophic lateral sclerosis with COVID-19. Muscle Nerve 64(4):E18–E20. https://doi.org/10.1002/mus.27373
Garcez FB, Aliberti MJR, Poco PCE, Hiratsuka M, Takahashi SF, Coelho VA, Salotto DB, Moreira MLV, Jacob-Filho W, Avelino-Silva TJ (2020) Delirium and adverse outcomes in hospitalized patients with COVID-19. J Am Geriatr Soc 68(11):2440–2446. https://doi.org/10.1111/jgs.16803
Garg RK, Paliwal VK, Gupta A (2021) Encephalopathy in patients with COVID-19: a review. J Med Virol 93(1):206–222. https://doi.org/10.1002/jmv.26207
Gemelli Against C-P-ACSG (2020) Post-COVID-19 global health strategies: the need for an interdisciplinary approach. Aging Clin Exp Res 32(8):1613–1620. https://doi.org/10.1007/s40520-020-01616-x
Glasmacher SA, Larraz J, Mehta AR, Kearns PKA, Wong M, Newton J, Davenport R, Gorrie G, Morrison I, Carod Artal J, Chandran S, Pal S, Consortium C-M (2021) The immediate impact of the COVID-19 pandemic on motor neuron disease services and mortality in Scotland. J Neurol 268(6):2038–2040. https://doi.org/10.1007/s00415-020-10207-9
Gomes I, Karmirian K, Oliveira JT, Pedrosa C, Mendes MA, Rosman FC, Chimelli L, Rehen S (2021) SARS-CoV-2 infection of the central nervous system in a 14-month-old child: a case report of a complete autopsy. Lancet Reg Health Am 2:100046. https://doi.org/10.1016/j.lana.2021.100046
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202(3):415–424. https://doi.org/10.1084/jem.20050828
Guidance on “Long COVID” as a Disability Under the ADA, Section 504, and Section 1557 (2021). Accessed 13 Dec 2021
Haehner A, Hummel T, Reichmann H (2011) Olfactory loss in Parkinson’s disease. Parkinsons Dis 2011:450939. https://doi.org/10.4061/2011/450939
Haji Akhoundi F, Sahraian MA, Naser Moghadasi A (2020) Neuropsychiatric and cognitive effects of the COVID-19 outbreak on multiple sclerosis patients. Mult Scler Relat Disord 41:102164. https://doi.org/10.1016/j.msard.2020.102164
Harb AA, Chen R, Chase HS, Natarajan K, Noble JM (2021) Clinical features and outcomes of patients with dementia compared to an aging cohort hospitalized during the initial New York City COVID-19 wave. J Alzheimers Dis 81(2):679–690. https://doi.org/10.3233/JAD-210050
Hariyanto TI, Putri C, Arisa J, Situmeang RFV, Kurniawan A (2021) Dementia and outcomes from coronavirus disease 2019 (COVID-19) pneumonia: a systematic review and meta-analysis. Arch Gerontol Geriatr 93:104299. https://doi.org/10.1016/j.archger.2020.104299
Hayley S, Sun H (2021) Neuroimmune multi-hit perspective of coronaviral infection. J Neuroinflammation 18(1):231. https://doi.org/10.1186/s12974-021-02282-0
Heneka MT, Golenbock D, Latz E, Morgan D, Brown R (2020) Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res Ther 12(1):69. https://doi.org/10.1186/s13195-020-00640-3
Hippisley-Cox J, Coupland CA, Mehta N, Keogh RH, Diaz-Ordaz K, Khunti K, Lyons RA, Kee F, Sheikh A, Rahman S, Valabhji J, Harrison EM, Sellen P, Haq N, Semple MG, Johnson PWM, Hayward A, Nguyen-Van-Tam JS (2021) Risk prediction of covid-19 related death and hospital admission in adults after covid-19 vaccination: national prospective cohort study. BMJ 374:n2244. https://doi.org/10.1136/bmj.n2244
Hirschbuhl K, Dintner S, Beer M, Wylezich C, Schlegel J, Delbridge C, Borcherding L, Lippert J, Schiele S, Muller G, Moiraki D, Spring O, Wittmann M, Kling E, Braun G, Kroncke T, Claus R, Markl B, Schaller T (2021) Viral mapping in COVID-19 deceased in the Augsburg autopsy series of the first wave: a multiorgan and multimethodological approach. PLoS ONE 16(7):e0254872. https://doi.org/10.1371/journal.pone.0254872
Hoffman LA, Vilensky JA (2017) Encephalitis lethargica: 100 years after the epidemic. Brain 140(8):2246–2251. https://doi.org/10.1093/brain/awx177
Horn A, Krist L, Lieb W, Montellano FA, Kohls M, Haas K, Gelbrich G, Bolay-Gehrig SJ, Morbach C, Reese JP, Stork S, Fricke J, Zoller T, Schmidt S, Triller P, Kretzler L, Ronnefarth M, Von Kalle C, Willich SN, Kurth F, Steinbeis F, Witzenrath M, Bahmer T, Hermes A, Krawczak M, Reinke L, Maetzler C, Franzenburg J, Enderle J, Flinspach A, Vehreschild J, Schons M, Illig T, Anton G, Ungethum K, Finkenberg BC, Gehrig MT, Savaskan N, Heuschmann PU, Keil T, Schreiber S (2021) Long-term health sequelae and quality of life at least 6 months after infection with SARS-CoV-2: design and rationale of the COVIDOM-study as part of the NAPKON population-based cohort platform (POP). Infection 49(6):1277–1287. https://doi.org/10.1007/s15010-021-01707-5
Hu C, Chen C, Dong XP (2021) Impact of COVID-19 pandemic on patients with neurodegenerative diseases. Front Aging Neurosci 13:664965. https://doi.org/10.3389/fnagi.2021.664965
Hu Y, Yang H, Hou C, Chen W, Zhang H, Ying Z, Hu Y, Sun Y, Qu Y, Feychting M, Valdimarsdottir U, Song H, Fang F (2022) COVID-19 related outcomes among individuals with neurodegenerative diseases: a cohort analysis in the UK biobank. BMC Neurol 22(1):15. https://doi.org/10.1186/s12883-021-02536-7
Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, Kang L, Guo L, Liu M, Zhou X, Luo J, Huang Z, Tu S, Zhao Y, Chen L, Xu D, Li Y, Li C, Peng L, Li Y, Xie W, Cui D, Shang L, Fan G, Xu J, Wang G, Wang Y, Zhong J, Wang C, Wang J, Zhang D, Cao B (2021) 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet 397(10270):220–232. https://doi.org/10.1016/S0140-6736(20)32656-8
Huber MK, Raichle C, Lingor P, Synofzik M, Borgmann S, Erber J, Tometten L, Rimili W, Dolff S, Wille K, Knauss S, Piepel C, Lanznaster J, Rieg S, Prasser F, Pilgram L, Spottke A, Klockgether T, Klein C, Hopfner F, Hoglinger GU (2021) Outcomes of SARS-CoV-2 infections in patients with neurodegenerative diseases in the LEOSS cohort. Mov Disord 36(4):791–793. https://doi.org/10.1002/mds.28554
Hull M, Parnes M, Jankovic J (2021) Increased incidence of functional (psychogenic) movement disorders in children and adults amid the COVID-19 pandemic: a cross-sectional study. Neurol Clin Pract 11(5):e686–e690. https://doi.org/10.1212/CPJ.0000000000001082
Kanberg N, Simren J, Eden A, Andersson LM, Nilsson S, Ashton NJ, Sundvall PD, Nellgard B, Blennow K, Zetterberg H, Gisslen M (2021) Neurochemical signs of astrocytic and neuronal injury in acute COVID-19 normalizes during long-term follow-up. EBioMedicine 70:103512. https://doi.org/10.1016/j.ebiom.2021.103512
Kanduc D (2021) From Anti-SARS-CoV-2 immune response to the cytokine storm via molecular mimicry. Antibodies (basel). https://doi.org/10.3390/antib10040036
Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, Barro C, Kappos L, Comabella M, Fazekas F, Petzold A, Blennow K, Zetterberg H, Kuhle J (2018) Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 14(10):577–589. https://doi.org/10.1038/s41582-018-0058-z
Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H (2020) The architecture of SARS-CoV-2 transcriptome. Cell 181(4):914–921. https://doi.org/10.1016/j.cell.2020.04.011
Kim R, Shin CW, Kim HJ, Jeon BS (2015) Postencephalitic parkinsonism responsive to a dopamine agonist: a case report. Parkinsonism Relat Disord 21(6):667–668. https://doi.org/10.1016/j.parkreldis.2015.03.018
Kitani-Morii F, Kasai T, Horiguchi G, Teramukai S, Ohmichi T, Shinomoto M, Fujino Y, Mizuno T (2021) Risk factors for neuropsychiatric symptoms in patients with Parkinson’s disease during COVID-19 pandemic in Japan. PLoS ONE 16(1):e0245864. https://doi.org/10.1371/journal.pone.0245864
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, Sheffield C-GG, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182(4):812–827. https://doi.org/10.1016/j.cell.2020.06.043
Lara B, Carnes A, Dakterzada F, Benitez I, Pinol-Ripoll G (2020) Neuropsychiatric symptoms and quality of life in Spanish patients with Alzheimer’s disease during the COVID-19 lockdown. Eur J Neurol 27(9):1744–1747. https://doi.org/10.1111/ene.14339
Leavy B, Hagstromer M, Conradsson DM, Franzen E (2021) Physical activity and perceived health in people with Parkinson disease during the first wave of Covid-19 pandemic: a cross-sectional study From Sweden. J Neurol Phys Ther 45(4):266–272. https://doi.org/10.1097/NPT.0000000000000372
Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, Zhao C, Zhang Q, Liu H, Nie L, Qin H, Wang M, Lu Q, Li X, Sun Q, Liu J, Zhang L, Li X, Huang W, Wang Y (2020) The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182(5):1284–1294. https://doi.org/10.1016/j.cell.2020.07.012
Limphaibool N, Iwanowski P, Holstad MJV, Kobylarek D, Kozubski W (2019) Infectious etiologies of Parkinsonism: pathomechanisms and clinical implications. Front Neurol 10:652. https://doi.org/10.3389/fneur.2019.00652
Lin DY, Gu Y, Wheeler B, Young H, Holloway S, Sunny SK, Moore Z, Zeng D (2022) Effectiveness of Covid-19 vaccines over a 9-month period in North Carolina. N Engl J Med. https://doi.org/10.1056/NEJMoa2117128
Liu N, Sun J, Wang X, Zhao M, Huang Q, Li H (2020a) The impact of dementia on the clinical outcome of COVID-19: a systematic review and meta-analysis. J Alzheimers Dis 78(4):1775–1782. https://doi.org/10.3233/JAD-201016
Liu S, Hossinger A, Heumuller SE, Hornberger A, Buravlova O, Konstantoulea K, Muller SA, Paulsen L, Rousseau F, Schymkowitz J, Lichtenthaler SF, Neumann M, Denner P, Vorberg IM (2021) Highly efficient intercellular spreading of protein misfolding mediated by viral ligand-receptor interactions. Nat Commun 12(1):5739. https://doi.org/10.1038/s41467-021-25855-2
Liu Y, Gayle AA, Wilder-Smith A, Rocklov J (2020b) The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med. https://doi.org/10.1093/jtm/taaa021
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD, Sacco C, Bertuzzi A, Sandri MT, Barco S, Humanitas C-TF (2020) Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res 191:9–14. https://doi.org/10.1016/j.thromres.2020.04.024
Lorusso D, Ray-Coquard I, Oaknin A, Banerjee S (2020) Clinical research disruption in the post-COVID-19 era: will the pandemic lead to change? ESMO Open 5(5):e000924. https://doi.org/10.1136/esmoopen-2020-000924
Luijten LWG, Leonhard SE, van der Eijk AA, Doets AY, Appeltshauser L, Arends S, Attarian S, Benedetti L, Briani C, Casasnovas C, Castellani F, Dardiotis E, Echaniz-Laguna A, Garssen MPJ, Harbo T, Huizinga R, Humm AM, Jellema K, van der Kooi AJ, Kuitwaard K, Kuntzer T, Kusunoki S, Lascano AM, Martinez-Hernandez E, Rinaldi S, Samijn JPA, Scheidegger O, Tsouni P, Vicino A, Visser LH, Walgaard C, Wang Y, Wirtz PW, Ripellino P, Jacobs BC, Consortium I (2021) Guillain-Barre syndrome after SARS-CoV-2 infection in an international prospective cohort study. Brain 144(11):3392–3404. https://doi.org/10.1093/brain/awab279
Mahammedi A, Saba L, Vagal A, Leali M, Rossi A, Gaskill M, Sengupta S, Zhang B, Carriero A, Bachir S, Crivelli P, Pasche A, Premi E, Padovani A, Gasparotti R (2020) Imaging of neurologic disease in hospitalized patients with COVID-19: an Italian multicenter retrospective observational Study. Radiology 297(2):E270–E273. https://doi.org/10.1148/radiol.2020201933
Mahapure KS, Prabhune AS, Chouvhan AV (2021) COVID-19-associated acute disseminated encephalomyelitis: a systematic review. Asian J Neurosurg 16(3):457–469. https://doi.org/10.4103/ajns.AJNS_406_20
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Li Y, Hu B (2020) Neurologic manifestations of hospitalized patients With Coronavirus Disease 2019 in Wuhan. China JAMA Neurol 77(6):683–690. https://doi.org/10.1001/jamaneurol.2020.1127
Matschke J, Lutgehetmann M, Hagel C, Sperhake JP, Schroder AS, Edler C, Mushumba H, Fitzek A, Allweiss L, Dandri M, Dottermusch M, Heinemann A, Pfefferle S, Schwabenland M, Sumner Magruder D, Bonn S, Prinz M, Gerloff C, Puschel K, Krasemann S, Aepfelbacher M, Glatzel M (2020) Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol 19(11):919–929. https://doi.org/10.1016/S1474-4422(20)30308-2
McAlpine LS, Fesharaki-Zadeh A, Spudich S (2021) Coronavirus disease 2019 and neurodegenerative disease: what will the future bring? Curr Opin Psychiatry 34(2):177–185. https://doi.org/10.1097/YCO.0000000000000688
Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, Laue M, Schneider J, Brunink S, Greuel S, Lehmann M, Hassan O, Aschman T, Schumann E, Chua RL, Conrad C, Eils R, Stenzel W, Windgassen M, Rossler L, Goebel HH, Gelderblom HR, Martin H, Nitsche A, Schulz-Schaeffer WJ, Hakroush S, Winkler MS, Tampe B, Scheibe F, Kortvelyessy P, Reinhold D, Siegmund B, Kuhl AA, Elezkurtaj S, Horst D, Oesterhelweg L, Tsokos M, Ingold-Heppner B, Stadelmann C, Drosten C, Corman VM, Radbruch H, Heppner FL (2021) Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci 24(2):168–175. https://doi.org/10.1038/s41593-020-00758-5
Mendes A, Herrmann FR, Perivier S, Gold G, Graf CE, Zekry D (2021) Delirium in older patients with COVID-19: prevalence, risk factors, and clinical relevance. J Gerontol A Biol Sci Med Sci 76(8):e142–e146. https://doi.org/10.1093/gerona/glab039
Mendez-Guerrero A, Laespada-Garcia MI, Gomez-Grande A, Ruiz-Ortiz M, Blanco-Palmero VA, Azcarate-Diaz FJ, Rabano-Suarez P, Alvarez-Torres E, de Fuenmayor-Fernandez de la Hoz CP, Vega Perez D, Rodriguez-Montalban R, Perez-Rivilla A, Sayas Catalan J, Ramos-Gonzalez A, Gonzalez de la Aleja J (2020) Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 95 (15):e2109–e2118. https://doi.org/10.1212/WNL.0000000000010282
Misra S, Kolappa K, Prasad M, Radhakrishnan D, Thakur KT, Solomon T, Michael BD, Winkler AS, Beghi E, Guekht A, Pardo CA, Wood GK, Hsiang-Yi Chou S, Fink EL, Schmutzhard E, Kheradmand A, Hoo FK, Kumar A, Das A, Srivastava AK, Agarwal A, Dua T, Prasad K (2021) Frequency of neurologic manifestations in COVID-19: a systematic review and meta-analysis. Neurology 97(23):e2269–e2281. https://doi.org/10.1212/WNL.0000000000012930
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, Ueno M, Sakata H, Kondo K, Myose N, Nakao A, Takeda M, Haro H, Inoue O, Suzuki-Inoue K, Kubokawa K, Ogihara S, Sasaki T, Kinouchi H, Kojin H, Ito M, Onishi H, Shimizu T, Sasaki Y, Enomoto N, Ishihara H, Furuya S, Yamamoto T, Shimada S (2020) A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis 94:55–58. https://doi.org/10.1016/j.ijid.2020.03.062
Muhammed L, Baheerathan A, Cao M, Leite MI, Viegas S (2021) MuSK antibody-associated myasthenia gravis with SARS-CoV-2 infection: a case report. Ann Intern Med 174(6):872–873. https://doi.org/10.7326/L20-1298
Mulder J, Feresiadou A, Fallmar D, Frithiof R, Virhammar J, Rasmusson A, Rostami E, Kumlien E, Cunningham JL (2021) Autoimmune encephalitis presenting with malignant catatonia in a 40-year-old male patient with COVID-19. Am J Psychiatry 178(6):485–489. https://doi.org/10.1176/appi.ajp.2020.20081236
Musson LS, Collins A, Opie-Martin S, Bredin A, Hobson EV, Barkhouse E, Coulson MC, Stavroulakis T, Gould RL, Al-Chalabi A, McDermott CJ (2022) Impact of the covid-19 pandemic on amyotrophic lateral sclerosis care in the UK. Amyotroph Lateral Scler Frontotemporal Degener. https://doi.org/10.1080/21678421.2022.2040533
Nasserie T, Hittle M, Goodman SN (2021) Assessment of the frequency and variety of persistent symptoms among patients with COVID-19: a systematic review. JAMA Netw Open 4(5):e2111417. https://doi.org/10.1001/jamanetworkopen.2021.11417
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82(15):7264–7275. https://doi.org/10.1128/JVI.00737-08
Neumann B, Schmidbauer ML, Dimitriadis K, Otto S, Knier B, Niesen WD, Hosp JA, Gunther A, Lindemann S, Nagy G, Steinberg T, Linker RA, Hemmer B, Bosel J, Pandemic, the Isg (2020) Cerebrospinal fluid findings in COVID-19 patients with neurological symptoms. J Neurol Sci 418:117090. https://doi.org/10.1016/j.jns.2020.117090
NICE R and SIGN (2022) COVID-19 rapid guideline: managing the long-term effects of COVID-19
Nishioku T, Dohgu S, Takata F, Eto T, Ishikawa N, Kodama KB, Nakagawa S, Yamauchi A, Kataoka Y (2009) Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol 29(3):309–316. https://doi.org/10.1007/s10571-008-9322-x
Oaklander AL, Mills AJ, Kelley M, Toran LS, Smith B, Dalakas MC, Nath A (2022) Peripheral neuropathy evaluations of patients with prolonged long COVID. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000001146
Parsons T, Banks S, Bae C, Gelber J, Alahmadi H, Tichauer M (2020) COVID-19-associated acute disseminated encephalomyelitis (ADEM). J Neurol 267(10):2799–2802. https://doi.org/10.1007/s00415-020-09951-9
Paterson RW, Benjamin LA, Mehta PR, Brown RL, Athauda D, Ashton NJ, Leckey CA, Ziff OJ, Heaney J, Heslegrave AJ, Benedet AL, Blennow K, Checkley AM, Houlihan CF, Mummery CJ, Lunn MP, Manji H, Zandi MS, Keddie S, Chou M, Vinayan Changaradil D, Solomon T, Keshavan A, Barker S, Jager HR, Carletti F, Simister R, Werring DJ, Spyer MJ, Nastouli E, Gauthier S, Rosa-Neto P, Group UQSC-BS, Zetterberg H, Schott JM (2021) Serum and cerebrospinal fluid biomarker profiles in acute SARS-CoV-2-associated neurological syndromes. Brain Commun 3(3):fcab099. https://doi.org/10.1093/braincomms/fcab099
Post-COVID Conditions (2021) https://www.cdc.gov/coronavirus/2019-ncov/long-term-effects/index.html. Accessed 13 Dec 2021
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B (2020) COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology 296(2):E119–E120. https://doi.org/10.1148/radiol.2020201187
Prosperini L, Tortorella C, Haggiag S, Ruggieri S, Galgani S, Gasperini C (2022) Determinants of COVID-19-related lethality in multiple sclerosis: a meta-regression of observational studies. J Neurol. https://doi.org/10.1007/s00415-021-10951-6
Prudencio M, Erben Y, Marquez CP, Jansen-West KR, Franco-Mesa C, Heckman MG, White LJ, Dunmore JA, Cook CN, Lilley MT, Song Y, Harlow CF, Oskarsson B, Nicholson KA, Wszolek ZK, Hickson LJ, O’Horo JC, Hoyne JB, Gendron TF, Meschia JF, Petrucelli L (2021) Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abi7643
Radmanesh A, Raz E, Zan E, Derman A, Kaminetzky M (2020) Brain imaging use and findings in COVID-19: a single academic center experience in the epicenter of disease in the United States. AJNR Am J Neuroradiol 41(7):1179–1183. https://doi.org/10.3174/ajnr.A6610
Ramani A, Muller L, Ostermann PN, Gabriel E, Abida-Islam P, Muller-Schiffmann A, Mariappan A, Goureau O, Gruell H, Walker A, Andree M, Hauka S, Houwaart T, Dilthey A, Wohlgemuth K, Omran H, Klein F, Wieczorek D, Adams O, Timm J, Korth C, Schaal H, Gopalakrishnan J (2020) SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J 39(20):e106230. https://doi.org/10.15252/embj.2020106230
Ramiz L, Contrand B, Rojas Castro MY, Dupuy M, Lu L, Sztal-Kutas C, Lagarde E (2021) A longitudinal study of mental health before and during COVID-19 lockdown in the French population. Global Health 17(1):29. https://doi.org/10.1186/s12992-021-00682-8
Rebora P, Rozzini R, Bianchetti A, Blangiardo P, Marchegiani A, Piazzoli A, Mazzeo F, Cesaroni G, Chizzoli A, Guerini F, Bonfanti P, Morandi A, Faraci B, Gentile S, Bna C, Savelli G, Citerio G, Valsecchi MG, Mazzola P, Bellelli G, CoVi DLT (2021) Delirium in patients with SARS-CoV-2 infection: a multicenter study. J Am Geriatr Soc 69(2):293–299. https://doi.org/10.1111/jgs.16969
Restivo DA, Centonze D, Alesina A, Marchese-Ragona R (2020) Myasthenia gravis associated with SARS-CoV-2 infection. Ann Intern Med 173(12):1027–1028. https://doi.org/10.7326/L20-0845
Richter D, Scherbaum R, Bartig D, Gold R, Krogias C, Tonges L (2021) Analysis of nationwide multimodal complex treatment and drug pump therapy in Parkinson’s disease in times of COVID-19 pandemic in Germany. Parkinsonism Relat Disord 85:109–113. https://doi.org/10.1016/j.parkreldis.2021.03.006
Rifino N, Censori B, Agazzi E, Alimonti D, Bonito V, Camera G, Conti MZ, Foresti C, Frigeni B, Gerevini S, Grimoldi M, La Gioia S, Partziguian T, Quadri S, Riva R, Servalli MC, Sgarzi M, Storti B, Vedovello M, Venturelli E, Vigano M, Callegaro A, Arosio M, Sessa M (2021) Neurologic manifestations in 1760 COVID-19 patients admitted to Papa Giovanni XXIII Hospital, Bergamo. Italy J Neurol 268(7):2331–2338. https://doi.org/10.1007/s00415-020-10251-5
Romero-Sanchez CM, Diaz-Maroto I, Fernandez-Diaz E, Sanchez-Larsen A, Layos-Romero A, Garcia-Garcia J, Gonzalez E, Redondo-Penas I, Perona-Moratalla AB, Del Valle-Perez JA, Gracia-Gil J, Rojas-Bartolome L, Feria-Vilar I, Monteagudo M, Palao M, Palazon-Garcia E, Alcahut-Rodriguez C, Sopelana-Garay D, Moreno Y, Ahmad J, Segura T (2020) Neurologic manifestations in hospitalized patients with COVID-19: The ALBACOVID registry. Neurology 95(8):e1060–e1070. https://doi.org/10.1212/WNL.0000000000009937
Rosenberg ES, Dorabawila V, Easton D, Bauer UE, Kumar J, Hoen R, Hoefer D, Wu M, Lutterloh E, Conroy MB, Greene D, Zucker HA (2022) Covid-19 vaccine effectiveness in New York State. N Engl J Med 386(2):116–127. https://doi.org/10.1056/NEJMoa2116063
Ross Russell AL, Hardwick M, Jeyanantham A, White LM, Deb S, Burnside G, Joy HM, Smith CJ, Pollak TA, Nicholson TR, Davies NWS, Manji H, Easton A, Ray S, Zandi MS, Coles JP, Menon DK, Varatharaj A, McCausland B, Ellul MA, Thomas N, Breen G, Keddie S, Lunn MP, Burn JPS, Quattrocchi G, Dixon L, Rice CM, Pengas G, Al-Shahi Salman R, Carson A, Joyce EM, Turner MR, Benjamin LA, Solomon T, Kneen R, Pett S, Thomas RH, Michael BD, Galea I (2021) Spectrum, risk factors and outcomes of neurological and psychiatric complications of COVID-19: a UK-wide cross-sectional surveillance study. Brain Commun 3(3):fcab168. https://doi.org/10.1093/braincomms/fcab168
Rozzini R, Bianchetti A, Mazzeo F, Cesaroni G, Bianchetti L, Trabucchi M (2020) Delirium: clinical presentation and outcomes in older COVID-19 patients. Front Psychiatry 11:586686. https://doi.org/10.3389/fpsyt.2020.586686
Salari M, Zaker Harofteh B, Etemadifar M, Sedaghat N, Nouri H (2021) Movement disorders associated with COVID-19. Parkinsons Dis 2021:3227753. https://doi.org/10.1155/2021/3227753
Salari M, Zali A, Ashrafi F, Etemadifar M, Sharma S, Hajizadeh N, Ashourizadeh H (2020) Incidence of anxiety in Parkinson’s disease during the Coronavirus Disease (COVID-19) pandemic. Mov Disord 35(7):1095–1096. https://doi.org/10.1002/mds.28116
Scherbaum R, Kwon EH, Richter D, Bartig D, Gold R, Krogias C, Tonges L (2021) Clinical profiles and mortality of COVID-19 inpatients with Parkinson’s disease in Germany. Mov Disord 36(5):1049–1057. https://doi.org/10.1002/mds.28586
Shahali H, Ghasemi A, Farahani RH, Nezami Asl A, Hazrati E (2021) Acute transverse myelitis after SARS-CoV-2 infection: a rare complicated case of rapid onset paraplegia. J Neurovirol 27(2):354–358. https://doi.org/10.1007/s13365-021-00957-1
Shalash A, Roushdy T, Essam M, Fathy M, Dawood NL, Abushady EM, Elrassas H, Helmi A, Hamid E (2020) Mental health, physical activity, and quality of life in Parkinson’s disease during COVID-19 pandemic. Mov Disord 35(7):1097–1099. https://doi.org/10.1002/mds.28134
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807):221–224. https://doi.org/10.1038/s41586-020-2179-y
Shi Y, Guo M, Yang W, Liu S, Zhu B, Yang L, Yang C, Liu C (2021) Is SARS-CoV-2 vaccination safe and effective for elderly individuals with neurodegenerative diseases? Expert Rev Vaccines 20(4):375–383. https://doi.org/10.1080/14760584.2021.1911653
Snider BJ, Holtzman DM (2021) Effects of COVID-19 on preclinical and clinical research in neurology: examples from research on neurodegeneration and Alzheimer’s disease. Neuron 109(20):3199–3202. https://doi.org/10.1016/j.neuron.2021.08.026
Strafella C, Caputo V, Termine A, Barati S, Gambardella S, Borgiani P, Caltagirone C, Novelli G, Giardina E, Cascella R (2020) Analysis of ACE2 genetic variability among populations highlights a possible link with COVID-19-related neurological complications. Genes (basel). https://doi.org/10.3390/genes11070741
Tahira AC, Verjovski-Almeida S, Ferreira ST (2021) Dementia is an age-independent risk factor for severity and death in COVID-19 inpatients. Alzheimers Dement. https://doi.org/10.1002/alz.12352
Takahashi M, Yamada T, Nakajima S, Nakajima K, Yamamoto T, Okada H (1995) The substantia nigra is a major target for neurovirulent influenza A virus. J Exp Med 181(6):2161–2169. https://doi.org/10.1084/jem.181.6.2161
Tang KT, Hsu BC, Chen DY (2021) Autoimmune and rheumatic manifestations associated with COVID-19 in adults: an updated systematic review. Front Immunol 12:645013. https://doi.org/10.3389/fimmu.2021.645013
Thompson MG, Stenehjem E, Grannis S, Ball SW, Naleway AL, Ong TC, DeSilva MB, Natarajan K, Bozio CH, Lewis N, Dascomb K, Dixon BE, Birch RJ, Irving SA, Rao S, Kharbanda E, Han J, Reynolds S, Goddard K, Grisel N, Fadel WF, Levy ME, Ferdinands J, Fireman B, Arndorfer J, Valvi NR, Rowley EA, Patel P, Zerbo O, Griggs EP, Porter RM, Demarco M, Blanton L, Steffens A, Zhuang Y, Olson N, Barron M, Shifflett P, Schrag SJ, Verani JR, Fry A, Gaglani M, Azziz-Baumgartner E, Klein NP (2021) Effectiveness of Covid-19 vaccines in ambulatory and inpatient care settings. N Engl J Med 385(15):1355–1371. https://doi.org/10.1056/NEJMoa2110362
Thygesen LC, Moller SP, Ersboll AK, Santini ZI, Nielsen MBD, Gronbaek MK, Ekholm O (2021) Decreasing mental well-being during the COVID-19 pandemic: a longitudinal study among Danes before and during the pandemic. J Psychiatr Res 144:151–157. https://doi.org/10.1016/j.jpsychires.2021.09.035
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, Franciotta D, Baldanti F, Daturi R, Postorino P, Cavallini A, Micieli G (2020) Guillain-Barre syndrome associated with SARS-CoV-2. N Engl J Med 382(26):2574–2576. https://doi.org/10.1056/NEJMc2009191
Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, Sultan M, Easton A, Breen G, Zandi M, Coles JP, Manji H, Al-Shahi Salman R, Menon DK, Nicholson TR, Benjamin LA, Carson A, Smith C, Turner MR, Solomon T, Kneen R, Pett SL, Galea I, Thomas RH, Michael BD, CoroNerve Study G (2020) Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry 7(10):875–882. https://doi.org/10.1016/S2215-0366(20)30287-X
Virhammar J, Naas A, Fallmar D, Cunningham JL, Klang A, Ashton NJ, Jackmann S, Westman G, Frithiof R, Blennow K, Zetterberg H, Kumlien E, Rostami E (2021) Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur J Neurol 28(10):3324–3331. https://doi.org/10.1111/ene.14703
von Bartheld CS, Hagen MM, Butowt R (2020) Prevalence of chemosensory dysfunction in COVID-19 patients: a systematic review and meta-analysis reveals significant ethnic differences. medRxiv. https://doi.org/10.1101/2020.06.15.20132134
Watson CJ, Thomas RH, Solomon T, Michael BD, Nicholson TR, Pollak TA (2021) COVID-19 and psychosis risk: real or delusional concern? Neurosci Lett 741:135491. https://doi.org/10.1016/j.neulet.2020.135491
Wei G, Diehl-Schmid J, Matias-Guiu JA, Pijnenburg Y, Landin-Romero R, Bogaardt H, Piguet O, Kumfor F (2022) The effects of the COVID-19 pandemic on neuropsychiatric symptoms in dementia and carer mental health: an international multicentre study. Sci Rep 12(1):2418. https://doi.org/10.1038/s41598-022-05687-w
WHO Coronavirus (COVID-19) Dashboard (2022) https://covid19.who.int/. Accessed 13 Apr 2022
Widmann CN, Heneka MT (2014) Long-term cerebral consequences of sepsis. Lancet Neurol 13(6):630–636. https://doi.org/10.1016/S1474-4422(14)70017-1
Williams FMK, Freidin MB, Mangino M, Couvreur S, Visconti A, Bowyer RCE, Le Roy CI, Falchi M, Mompeo O, Sudre C, Davies R, Hammond C, Menni C, Steves CJ, Spector TD (2020) Self-reported symptoms of COVID-19, including symptoms most predictive of SARS-CoV-2 infection, are heritable twin res. Hum Genet 23(6):316–321. https://doi.org/10.1017/thg.2020.85
Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, Curtis HJ, Mehrkar A, Evans D, Inglesby P, Cockburn J, McDonald HI, MacKenna B, Tomlinson L, Douglas IJ, Rentsch CT, Mathur R, Wong AYS, Grieve R, Harrison D, Forbes H, Schultze A, Croker R, Parry J, Hester F, Harper S, Perera R, Evans SJW, Smeeth L, Goldacre B (2020) Factors associated with COVID-19-related death using OpenSAFELY. Nature 584(7821):430–436. https://doi.org/10.1038/s41586-020-2521-4
Wolff AW, Haller B, Demleitner AF, Westenberg E, Lingor P (2022) Impact of the COVID-19 pandemic on patients with Parkinson’s disease from the perspective of treating physicians-a nationwide cross-sectional study. Brain Sci. https://doi.org/10.3390/brainsci12030353
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263. https://doi.org/10.1126/science.abb2507
Wu J, Deng W, Li S, Yang X (2021) Advances in research on ACE2 as a receptor for 2019-nCoV. Cell Mol Life Sci 78(2):531–544. https://doi.org/10.1007/s00018-020-03611-x
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107(6):1482–1494. https://doi.org/10.1111/j.1471-4159.2008.05723.x
Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, Tang X, Zhu J, Zhao Z, Jaffre F, Zhang T, Kim TW, Harschnitz O, Redmond D, Houghton S, Liu C, Naji A, Ciceri G, Guttikonda S, Bram Y, Nguyen DT, Cioffi M, Chandar V, Hoagland DA, Huang Y, Xiang J, Wang H, Lyden D, Borczuk A, Chen HJ, Studer L, Pan FC, Ho DD, tenOever BR, Evans T, Schwartz RE, Chen S (2020) A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell 27(1):125–136. https://doi.org/10.1016/j.stem.2020.06.015
Zanella I, Zacchi E, Piva S, Filosto M, Beligni G, Alaverdian D, Amitrano S, Fava F, Baldassarri M, Frullanti E, Meloni I, Renieri A, Study G-CM, Group GS, Castelli F, Quiros-Roldan E (2021) C9orf72 intermediate repeats confer genetic risk for severe COVID-19 pneumonia independently of age. Int J Mol Sci. https://doi.org/10.3390/ijms22136991
Zhang Q, Schultz JL, Aldridge GM, Simmering JE, Kim Y, Ogilvie AC, Narayanan NS (2021a) COVID-19 case fatality and Alzheimer’s disease. J Alzheimers Dis 84(4):1447–1452. https://doi.org/10.3233/JAD-215161
Zhang Q, Schultz JL, Aldridge GM, Simmering JE, Kim Y, Ogilvie AC, Narayanan NS (2021b) COVID-19 case fatality and Alzheimer’s disease. J Alzheimers Dis. https://doi.org/10.3233/JAD-215161
Zhang Q, Schultz JL, Aldridge GM, Simmering JE, Narayanan NS (2020) Coronavirus disease 2019 case fatality and Parkinson’s disease. Mov Disord 35(11):1914–1915. https://doi.org/10.1002/mds.28325
Zhou L, Zhang M, Wang J, Gao J (2020a) Sars-Cov-2: underestimated damage to nervous system. Travel Med Infect Dis 36:101642. https://doi.org/10.1016/j.tmaid.2020.101642
Zhou S, Jones-Lopez EC, Soneji DJ, Azevedo CJ, Patel VR (2020b) Myelin oligodendrocyte glycoprotein antibody-associated optic neuritis and myelitis in COVID-19. J Neuroophthalmol 40(3):398–402. https://doi.org/10.1097/WNO.0000000000001049
Zhou Y, Han T, Chen J, Hou C, Hua L, He S, Guo Y, Zhang S, Wang Y, Yuan J, Zhao C, Zhang J, Jia Q, Zuo X, Li J, Wang L, Cao Q, Jia E (2020c) Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin Transl Sci 13(6):1077–1086. https://doi.org/10.1111/cts.12805
Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S (2020) Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurol 77(8):1018–1027. https://doi.org/10.1001/jamaneurol.2020.2065
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial or non-financial interests related to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lingor, P., Demleitner, A.F., Wolff, A.W. et al. SARS-CoV-2 and neurodegenerative diseases: what we know and what we don’t. J Neural Transm 129, 1155–1167 (2022). https://doi.org/10.1007/s00702-022-02500-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02500-w