
Abstract
Parkinson’s disease is the second most common neurological disorder marked by characteristic poverty and dysfunction in movement. There are many mechanisms and factors which have been postulated to be associated with the neurodegenerative pathway(s) resulting in distinctive loss of neurons in the substantia nigra. Subsequently, the neuropathology is more widespread and exhibited in other areas of the brain, and enteric nervous system. Aggregates of misfolded α-synuclein or Lewy bodies are the hallmark of the illness and appear to be central in the whole cascade of cell destruction. There are many processes implicated in neuronal destruction including: oxidative stress, excitotoxicity, mitochondrial dysfunction, an imbalance in protein homeostasis and neuroinflammation. Interesting, inflammation induced by pathogens (including, bacteria and viruses) has been associated in the pathogenesis of the disease. Bacteria such as Helicobacter pylori and Helicobacter suis appear to colonise the gut, and elicit systemic immune responses, which is them transmitted via the gut-axis to the brain, where cytotoxic cytokines induce neuroinflammation and cell death. This conforms to the bottom–top hypothesis proposed by Braak. The gut is also implicated in two other theories postulated in the development and progression of the disorder, namely, the top–down and the threshold. There is a possibility that these theories may be inter-linked and operate together to certain degree. Ultimately specific trigger factors or conditions may govern the occurrences of these processes in genetically predisposed individuals. Nevertheless, the importance of pathogen-related gut infections cannot be overlooked, since it can result in dysbiosis of gut microbes, which may orchestrate α-synuclein pathology and eventually cell destruction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The neurodegenerative disorder, Parkinson’s disease is classically marked by progressive motor deficits coupled with the characteristic presence of neuropathologic features such as Lewy bodies (LB) and loss of melanin containing dopaminergic neurons primarily in the ventrolateral tier of the substantia nigra (SN) pars compacta. Despite the flurry of potential candidates, the origin of the disorder largely remains obscure. The major aspirants implicated include, increasing age, a genetic predilection and a possible neurotoxin. Subsequently, they may initiate or contribute in the irregular folding of α-synuclein protein, leading to its accumulation and generation of LB structures in the nigral neurons and other affected cells. These aberrant α-synuclein accumulations manifest into rogue proteins, which produce chaos and cellular destruction in its wake. It employs cytotoxic processes such as, neuroinflammation, oxidative stress, excitotoxicity. This, may contribute to, malfunction in mitochondria (Schapira et al. 1989), iron dyshomeostasis (Riederer et al. 1989), impaired operation of protein autophagic system (Sian-Hulsmann and Riederer 2020) and reduced dopamine transmission. This is coupled with a compromised cellular antioxidant defence, glutathione (Sian et al. 1991) thereby making the dopaminergic cells vulnerable to the onslaught of pathological mechanism(s).
It appears highly likely that as opposed to one causal agent, that instead these mechanisms work in collaboration to initiate the cellular degeneration. Thus, the pathogenesis of the illness is probably multifactorial with an assortment of possible permutations of possible causal factors which operate in concert to execute the pathway resulting in neuronal destruction in individuals with a predilection (Pang et al. 2019). Consequently, the different factors involved may contribute to the PD subtypes. Previously, based on the motor deficits PD subtypes were categorised in two main groups, tremor dominant and non-tremor dominant (postural instability with gait difficulty) (Nutt 2016). However, cognitive/psychiatric characteristics may also be employed to distinguish between the subtypes (Campbell et al. 2020). Thus, using multi-domain latent class analysis, three major subtypes for PD have been categorised namely, motor only, cognitive and motor, and psychiatric and motor (Campbell et al. 2020). Importantly, the variability in the subtypes may be primarily due to the occurrence of different pathomechanisms, this needs to be carefully considered particularly in the advocation of therapeutic strategies targeted towards neuroprotection or disease modification. Nevertheless, in the terminating stages of the illness, it appears that the clinical features and the neuropathological changes merge in the various PD subtypes. This notion is demonstrated by the similarity in the staging of LB pathology in the PD subtypes (De Pablo-Fernandez et al. 2019).
Interestingly, the spotlight has been focussed on neuroinflammation as a budding process in eliciting cell death in PD (Hirsch et al. 2012). The occurrence of inflammation was first suggested by the microgliosis found in the SN in PD (McGeer et al. 1988). This is a significant finding since microglia are vital immune cells that are elite members of the central nervous system defence and more importantly, they have the authority to coordinate inflammatory processes. Apparently, the function of the microglia appears to be ambivalent and is determined by the local cellular environment and is thus pathological dependent. Therefore, under physiological conditions it serves a host of protective cellular functions including, control of neuronal excitability and neuronal support. However, the pathological state may alter the microglial cellular environment resulting in a switch of microglial functions and proliferation to a more sinister role (Gomez-Nicola and Perry 2014). Indeed, in the diseased state it may execute the release of molecular regulators such as, cytokines and chemokines that control inflammation (Kettenmann et al. 2011).
In addition, powerful pro-inflammatory cytokines (such as, interleukin 1β) may be released from the interaction of reactive microglia with Toll-like receptor (Lehnardt 2009). This association with the Toll-like receptors may facilitate innate and adaptive immunity. Additionally, cytotoxic cytokine molecules (particularly interferons and interleukins) can evoke cellular destruction via generation of reactive free radical species, which subsequently trigger the cascade of oxidative stress resulting in neuronal destruction. In fact, raised levels of cytokines such as, tumour necrosis factor- α, interleukins-1β,6 and interferon gamma have been documented in PD brains (Mount et al. 2007; Chao et al. 2014).
It has been suggested that the release of oxidised or nitrated α-synuclein (modified) form present in LB from dying dopaminergic neurons, may release superoxide and nitric oxide, these may release toxic free radicals’ species that finally trigger immune reactions (Gao et al. 2008a). Indeed, the modified version of α-synuclein has been implicated to be key in microgliosis (Lee et al. 2008). Thereby endorsing the involvement of α-synuclein in the intricate pathomechanism of the disease. Additionally, the elevated nigral iron levels in PD may also have the propensity to activate microgliosis and release of oxidative species (Liu et al. 2017a).
The neuropathological changes in incidental Lewy body disease represent early changes in the asymptomatic phase of PD (Gibb and Lees 1988). Interestingly, the only changes in nigral neurons observed were, reduction of glutathione (Dexter et al. 1994), loss of dopaminergic neurons and the presence of LB. This suggests the operation of some active oxidative stress processes early in the pathogenesis, which contributes to the depletion of the antioxidant glutathione. The destruction of the dopamine cells may also evoke protective immune responses. Therefore, although neuroinflammation may not be associated to the initial onslaught of endogenous characteristic dopamine neuronal destructive processes, nevertheless it has the compelling ability in its repertoire to operate as an epiphenomenon.
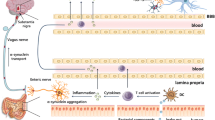
By virtue of their ability to trigger immune responses, pathogens such as bacteria and viruses may provoke neuronal destruction. The human gut has the large population of microbes, that serve vital physiological functions. However, they can adopt an ominous function and recent studies have shown an association between gut microbiota and neurological disorders (Gorle et al. 2021). In fact, there are reports suggestive of peripheral inflammation which is prompted by bacterial derived inflammagens which consequently spreads to the brain (Vuuren et al. 2020). This may play a key role in the development of PD. It appears that these microbes can connect with the brain through a number of routes collectively called micobiome-gut-brain axis. These routes include; metabolic, endocrine, metabolic, neuronal and immunological. Bacteria have the propensity to affect the make-up of normal gut flora.
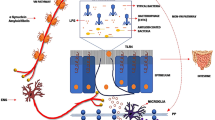
The Gram-negative bacteria, Helicobacter widely colonises the stomach and intestine. Particularly Helicobacter pylori (H. pylori), which is commonly associated with gastric and duodenal ulcers (Lanas and Chan 2017). It resides on the gastric lumen and can either increase or decrease the hydrochloric acid output in the gut. In the case of a high acid output, it is related to an elevation of gastrin, that results in antrum- predominant gastritis and results in duodenal ulcers. In contrast, a low acid output produces diffuse gastritis leading to gastric ulcers. The H. pylori infection is usually contracted in the early years of life and if left untreated it can develop into gastritis and ulcers. The gastritis is a direct consequence of chronic inflammation produced by the bacteria. The pathological abilities of this bacteria are not limited to gastric ulcers only and it has also been associated with an assortment of diseases including; neurological diseases (PD and Alzheimer’s disease), cardiovascular disease and anaemia. Interestingly, a meta-analysis study showed a high incidence of PD patients that were infected with H. pylori (Dardiotis et al. 2018). Additionally, PD patients infected with this bacterium exhibited improved clinical status after H. pylori antibiotic eradication treatment. This notion was exhibited by the lower UPDRS score (unified Parkinson’s disease rating scale). The remarkable ability of the bacterial infection to exacerbate the motor deficits or cause motor fluctuations, is chiefly related to the bacteria interfering with the absorption of the levodopa, which is the gold standard drug treatment of the disease (Camci and Oguz 2016). Thus, H. pylori eradication improves the bioavailability of levodopa thereby improving hypokinesia associated to the illness. Nevertheless, there is an alliance between H. pylori and PD, whereby it shares similar pathways linked to the pathogenesis of PD. The common denominator for the two appears to involve activation of the immune system.
Interestingly, McGee and colleagues (2018) proposed a hypothesis of mechanisms that the bacteria may orchestrate leading to PD. They suggested that H. pylori may produce a toxin, or/and effect gut microbiota and that the resulting inflammation caused by gut infection may cross the gut-brain axis to produce neuroinflammation in the brain. This is in accordance with notion reported by Przuntek and colleagues (2004), they proposed that the premotor phase may begin in the gastrointestinal tract spreading to SN and cortical areas via the spinal cord and brain stem. Perhaps a pathogen or some nutritional element in individuals with a genetic susceptibility for enteric disturbances would confer a vulnerability to developing PD (Przuntek et al. 2004). Additionally, McGee’s theory (2018) endorses Braak’s theory (‘bottom–up’), which suggests that the disease originates in the gut and subsequently the pathology spreads to the brain via the vagal innervation (Braak et al. 2006).
It is important to note that H. pylori is not the only pathogen exclusively associated to PD. Indeed, there is an array of bacteria associated with parkinsonism such as, Norcardia asteroids, Proteus mirabilis, Chlamydia pneumonia, Bordetella pertusiss, Streptococus pyogenes (Limphaibool et al. 2019). In addition, other pathogens such as viruses that also may evoke PD like features include, post-encephalitic virus, H5N1, Herpes simplex virus, West Nile, HIV, Hepatitis C (Limphaibool et al. 2019). Indeed, the human immunodeficiency virus (HIV) induces the immune system to evoke inflammatory processes resulting in α-synuclein misfolding, microgliosis in the mid brain, and a loss of dopamine transmission (Eugenin 2010). This loss of dopamine is suspected to be responsible for producing parkinsonian motor abnormalities observed in some of the HIV positive patients (Koutsilieri et al. 2002). Its seems that the infection and pathology may spread to the brain after a period of time has elapsed following the initial primary infection. Similarly, SARS-Cov-2 has also been related to promoting some neurological manifestations. The virus exerts cellular destruction via processes executed by the immune system and thromboembolism (Sian-Hulsmann and Riederer 2021). Indeed, the peripheral cytokines released by SARS-Cov-2 infection may the cross the blood brain barrier to induce cellular destruction by the release of reactive oxygen species and excitotoxic processes (Wu 2020).
Therefore, perhaps some pathogen such as a bacteria/virus or a toxin produced by it may stimulate immune processes that consequently trigger the onset of the disease. Indeed, neuroinflammation appears to be common feature shared by both infectious and neurodegenerative diseases. This concept is demonstrated by a study in which rats orally administered with curli protein made by Escherichia coli, amplified α-synuclein deposition in the gut plexi, hippocampus and striatum (Phillips et al. 2009). Subsequently, α-synuclein monomer or aggregates may traffic from the enteric nervous system to the brain via the vagus nerve. Infact patients who have had full truncal vagotomy are at a lower risk of developing PD, thereby confirming the vagal route (Liu et al. 2017b). Once in the brain the α-synuclein inclusions probably propagate in a prion like manner, whereby the main focus is the SN pars compacta in PD. However, as the disease progresses the pathological features spread to other areas of the brain.
Thus, it is conceivable that the inflammatory molecules released from H. pylori related gastritis may prompt the aggregation of α-synuclein. Indeed, expression of α-synuclein was directly linked to the degree of inflammation in the upper gastro intestinal (GI) tract (Stolzenberg et al. 2017). This concept is supported by the correlation between the α-synuclein in the neurites of the GI wall and mucosal inflammation associated to H. pylori infections in children. It has been suggested that the expression of α-synuclein may result as a physiological immune response (Stolzenberg et al. 2017). Subsequently, around this point other factors may come into play to finally activate cytotoxic processes that progress towards the development of the disease.
So, in the case of chronic gastritis as would be the case for untreated childhood H. pylori infections related gut infections, this may predispose towards PD (Barnum et al. 2012). The H. pylori virulence factors (such as production of cytotoxins) coupled with host characteristics (such as polymorphisms in host gene), and the external environment may lead to the cascade of inflammatory processes that finally define the vulnerability to the occurrences of different diseases (Termann and Morris 2014). This concept is demonstrated the greater risk of gastric cancer in individual that have polymorphism for the pro-inflammatory interleukin -1-β and interleukin-1 genes (Gao et al. 2008b). In addition, the (cytotoxin associated gene A) CagA strain has been reported to be virulent and confers a greater risk for the development of peptic ulcers (Yamaoka 2008). Additionally, apparently CagA expression is accompanied with presence of other virulence factors such as, oipA, babA and vacA. Translation of these finding to PD, would suggest that the virulence of the bacteria together with some genetic defaults and a host pro-inflammatory molecules exert a pivotal role in the pathophysiology.
Another gastric pathogen implicated in neurodegenerative diseases is, Helicobacter suis or H.suis. Recently, a high occurrence of H.suis was reported in PD patients (Gorle et al. 2021).
Interestingly, H.suis related gastric infections in mice resulted in leakage of gastrointestinal barrier coupled with brain inflammation (microgliosis) (Gorle et al. 2021). The bacteria probably induced gastrointestinal permeability and peripheral inflammation, resulting in the transport of immune molecules via the gut-brain axis, which may have distorted the blood cerebrospinal fluid barrier thus gaining access to the brain, and thereby supporting the bottom–up theory.
Although the bottom-up hypothesis offers a very plausible scenario in which H. pylori or H. suis can play a blossoming lead role, however the pattern of nigral neuronal degeneration postulated is not demonstrated in all cases. Thus, it has been challenged by other hypothesis, top–down (Foffani and Obeso 2018) and threshold theory (Engelender and Isacson 2017). Although, the top–down postulates a cortical pathogenesis beginning in the brain and spreading to the gut, nevertheless there is a possibility for it to coincide with the bottom-up (Leclair-Visonneau et al. 2019). The top–down hypothesis is demonstrated by experiments such as rats injected with the neurotoxin 6-hydroxydopamine into the brain, exhibited nigrostriatal lesion, a reduction nitric oxide synthase in enteric neurons and severe constipation (Colucci et al. 2012). Nitric oxide synthase is involved in the production of nitric oxide and interestingly both a depletion and an increase of it can exert pathological actions (Rochette et al. 2013). Indeed, nitric oxide synthase is implicated as one of the cheif culprits involved in the genesis of reactive nitrogen species that evoke oxidative stress, damage DNA and also perhaps cause the modification of α-synuclein resulting in the initiation of the cycle of cell destruction in PD. Therefore, this supports the feasibility of either/both the operation top–down and bottom-up mechanisms. Additionally, bilateral injection of adeno-associated virus- α-synuclein into the SN of rats resulted in effects in both the enteric nervous system and the gut flora (O’Donovan et al. 2019), thereby highlighting the importance of the gut-brain axis in the pathogenesis of the disease.
Alternatively, the threshold hypothesis for PD suggests a parallel degeneration occurring in the brain and peripheral nervous system (Engelender and Isacson 2017). Thus, the involvement of the gut in the pathophysiology of PD is feasible in all three theories and more importantly is endorsed by the common manifestation of gastrointestinal dysfunction in the disorder (Colucci et al. 2012). Perhaps, the route(s) of degeneration may be determined by a genetic component and the presence of a pathogen or other factors that eventually lead to the of onset for the illness.
H. pylori remains an enigmatic pathogen with many secrets yet to be revealed. In the absence of eradication treatment, H. pylori, can persist in the gut and produce a state of chronic inflammation resulting in the secretion of many its mediators including, pro-inflammatory cytokines (tumour necrosis factor- α, interleukins-1β,6 and interferon gamma), which are also reported to be elevated in PD (Chao et al. 2014). These pro-inflammatory molecules can gain entry to the brain via the gut-axis with the assistance of a “leaky” blood–brain barrier (Al-Bachari et al. 2020) where it can produce inflammation and related neuronal destruction.
Therefore, H. pylori and perhaps its “cousin”, H. suis represent formidable pathogens with the capacity to contribute to many disorders, including PD. By virtue of eliciting rogue immune reactions, they have the potential in collaboration with other features to unleash deadly pathomechanisms characteristic of the disorder. Indeed, factors such as, α-synuclein aggregates, inflammation in the enteric nervous system and brain, iron elevation in the SN and a disruption of gut flora by some pathogen may contrive to elicit the characteristic neurodegeneration exhibited in the disease (Vuuren et al. 2020). It therefore may be prudent to explore more carefully the pathways/processes elicited by these pathogens as it may lead to the aetiology of the illness or provide a better understanding of the underlying pathogenesis of PD. This may be vital to produce drugs that may halt or hinder the disease progression.
References
Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM (2020) Blood-brain barrier leakage is increased in Parkinson’s disease. Front Physiol. https://doi.org/10.3389/fphys.2020.593026
Barnum CJ, Tansey MG (2012) Neuroinflammation and Non-motor symptoms: the dark passenger of Parkinson’s disease? Curr Neurol Neurosci Rep 12(4):350–358
Braak H, de Vos RAI, Bohl J, Del Tredici K (2006) Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396(1):67–72
Çamcı G, Oğuz S (2016) Association between Parkinson’s Disease and Helicobacter Pylori. J Clin Neurol 12(2):147
Campbell MC, Myers PS, Weigand AJ, Foster ER, Cairns NJ, Jackson JJ, Lessov-Schlaggar CN, Perlmutter JS (2020) Parkinson disease clinical subtypes: key features & clinical milestones. Ann Clin Transl Neurol 7(8):1272–1283
Chao Y, Wong SC, Tan EK (2014) Evidence of inflammatory system involvement in Parkinson’s disease. BioMed Res Int 2014:1–9
Colucci M, Cervio M, Faniglione M, De Angelis S, Pajoro M, Levandis G, Tassorelli C, Blandini F, Feletti F, De Giorgio R, Dellabianca A, Tonini S, Tonini M (2012) Intestinal dysmotility and enteric neurochemical changes in a Parkinson’s disease rat model. Autonomic Neurosci 169(2):77–86
Dardiotis E, Tsouris Z, Mentis A-FA, Siokas V, Michalopoulou A, Sokratous M, Dastamani M, Bogdanos DP, Deretzi G, Kountouras J (2018) H. pylori and Parkinson’s disease: meta-analyses including clinical severity. Clin Neurol Neurosurg 175:16–24
De Pablo-Fernández E, Lees AJ, Holton JL, Warner TT (2019) Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 76(4):470
Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira AHV, Jenner P, Marsden CD (1994) Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol 35(1):38–44
Engelender S, Isacson O (2017) The threshold theory for Parkinson’s disease. Trends Neurosci 40(1):4–14
Eugenin E (2010) Novel mechanisms of central nervous system damage in HIV infection. HIV/AIDS - Res Palliative Care. https://doi.org/10.2147/HIV.S9186
Foffani G, Obeso JA (2018) A cortical pathogenic theory of Parkinson’s disease. Neuron 99(6):1116–1128
Gao H-M, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM-Y (2008a) Neuroinflammation and oxidation/nitration of -synuclein linked to dopaminergic neurodegeneration. J Neurosci 28(30):7687–7698
Gao L-B, Rao L, Wang Y-Y, Liang W-B, Li C, Xue H, Zhou B, Sun H, Li Y, Lv M-L, Du X-J, Zhang L (2008b) The association of interleukin-16 polymorphisms with IL-16 serum levels and risk of colorectal and gastric cancer. Carcinogenesis 30(2):295–299
Gibb WR, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 51(6):745–752
Gomez-Nicola D, Perry VH (2014) Microglial dynamics and role in the healthy and diseased brain. Neuroscientist 21(2):169–184
Gorlé N, Bauwens E, Haesebrouck F, Smet A, Vandenbroucke RE (2021) Helicobacter and the potential role in neurological disorders: there is more than Helicobacter pylori. Front Immunol. https://doi.org/10.3389/fimmu.2020.584165
Hirsch EC, Vyas S, Hunot S (2012) Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord 18:S210–S212
Kettenmann H, Hanisch U-K, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553
Koutsilieri E, Sopper S, Scheller C, ter Meulen V, Riederer P (2002) Parkinsonism in HIV dementia. J Neural Transm 109(5–6):767–775
Lanas A, Chan FKL (2017) Peptic ulcer disease. The Lancet 390(10094):613–624
Leclair-Visonneau L, Neunlist M, Derkinderen P, Lebouvier T (2019) The gut in Parkinson’s disease: Bottom-up, top-down, or neither? Neurogastroenterol & Motility. https://doi.org/10.1111/nmo.13777
Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, Lee S-J (2008) Assembly-dependent endocytosis and clearance of extracellular a-synuclein. Int J Biochem Cell Biol 40(9):1835–1849
Lehnardt S (2009) Innate immunity and neuroinflammation in the CNS: The role of microglia in Toll-like receptor-mediated neuronal injury. Glia. https://doi.org/10.1002/glia.20928
Limphaibool N, Iwanowski P, Holstad MJV, Kobylarek D, Kozubski W (2019) Infectious etiologies of Parkinsonism: pathomechanisms and clinical implications. Front Neurol. https://doi.org/10.3389/fneur.2019.00652
Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, Svenningsson P, Chen H, Wirdefeldt K (2017a) Vagotomy and Parkinson disease. Neurology 88(21):1996–2002
Liu Y, Ye Z, Li X, Anderson JL, Khan M, DaSilva D, Baron M, Wilson D, Bocoun V, Ivacic LC, Schrodi SJ, Smith JA (2017b) Genetic and functional associations with decreased anti-inflammatory tumor necrosis factor alpha induced protein 3 in macrophages from subjects with axial spondyloarthritis. Front Immunol. https://doi.org/10.3389/fimmu.2017.00860
McGee DJ, Lu X-H, Disbrow EA (2018) Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease. J Parkinson’s Dis 8(3):367–374
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38(8):1285–1285
Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, Anisman H, Hayley S, Park DS (2007) Involvement of interferon- in microglial-mediated loss of dopaminergic neurons. J Neurosci 27(12):3328–3337
Nutt JG (2016) Motor subtype in Parkinson’s disease: Different disorders or different stages of disease? Mov Disord 31(7):957–961
O’Donovan SM, Crowley EK, Brown JR-M, O’Sullivan O, O’Leary OF, Timmons S, Nolan YM, Clarke DJ, Hyland NP, Joyce SA, Sullivan AM, O’Neill C (2019) Nigral overexpression of α-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol & Motility. https://doi.org/10.1111/nmo.13726
Pang SY-Y, Ho PW-L, Liu H-F, Leung C-T, Li L, Chang EES, Ramsden DB, Ho S-L (2019) The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodegener. https://doi.org/10.1186/s40035-019-0165-9
Phillips RJ, Walter GC, Ringer BE, Higgs KM, Powley TL (2009) Alpha-synuclein immunopositive aggregates in the myenteric plexus of the aging Fischer 344 rat. Exp Neurol 220(1):109–119
Przuntek H, Muller Th, Riederer P (2004) Diagnostic staging of Parkinson?s disease: conceptual aspects. J Neural Transm 111(2):201–216
Riederer P, Sofic E, Rausch W-D, Schmidt B, Reynolds GP, Jellinger K, Youdim MBH (1989) Transition metals, ferritin, glutathione, and ascorbic acid in Parkinsonian brains. J Neurochem 52(2):515–520
Rochette L, Lorin J, Zeller M, Guilland J-C, Lorgis L, Cottin Y, Vergely C (2013) Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: possible therapeutic targets? Pharmacol Ther 140(3):239–257
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson’s disease. Lancet (london, England) 1(8649):1269
Sian J, Dexter DT, Jenner P et al (1991) Decrease in nigral reduced glutathione in Parkinson’s disease. Brit J Pharmacol 104:281
Sian-Hulsmann J, Riederer P (2020) The role of alpha-synuclein as ferrireductase in neurodegeneration associated with Parkinson’s disease. J Neural Transm 127(5):749–754
Sian-Hulsmann J, Riederer P (2021) The Nigral Coup in Parkinson’s Disease by α-Synuclein and its associated rebels. Cells 10(3):598
Stolzenberg E, Berry D, Yang D, Lee EY, Kroemer A, Kaufman S, Wong GCL, Oppenheim JJ, Sen S, Fishbein T, Bax A, Harris B, Barbut D, Zasloff MA (2017) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9(5):456–463
Testerman TL (2014) Beyond the stomach: An updated view of Helicobacter pyloripathogenesis, diagnosis, and treatment. World J Gastroenterol 20(36):12781
van Vuuren MJ, Nell TA, Carr JA, Kell DB, Pretorius E (2020) Iron dysregulation and inflammagens related to oral and gut health are central to the development of Parkinson’s disease. Biomolecules 11(1):30
Wu J (2020) Tackle the free radicals damage in COVID-19. Nitric Oxide 102:39–41
Yamaoka Y (2008) Roles of the plasticity regions of Helicobacter pylori in gastroduodenal pathogenesis. J Med Microbiol 57(5):545–553
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sian-Hülsmann, J. Wilful pathogens provoke a gut feeling in Parkinson’s disease. J Neural Transm 129, 557–562 (2022). https://doi.org/10.1007/s00702-021-02448-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-021-02448-3