
Abstract
Introduction
Traumatic axonal injury (TAI) is a condition defined as multiple, scattered, small hemorrhagic, and/or non-hemorrhagic lesions, alongside brain swelling, in a more confined white matter distribution on imaging studies, together with impaired axoplasmic transport, axonal swelling, and disconnection after traumatic brain injury (TBI). Ever since its description in the 1980s and the grading system by Adams et al., our understanding of the processes behind this entity has increased.
Methods
We performed a scoping systematic, narrative review by interrogating Ovid MEDLINE, Embase, and Google Scholar on the pathophysiology, biomarkers, and diagnostic tools of TAI patients until July 2020.
Results
We underline the misuse of the Adams classification on MRI without proper validation studies, and highlight the hiatus in the scientific literature and areas needing more research. In the past, the theory behind the pathophysiology relied on the inertial force exerted on the brain matter after severe TBI inducing a primary axotomy. This theory has now been partially abandoned in favor of a more refined theory involving biochemical processes such as protein cleavage and DNA breakdown, ultimately leading to an inflammation cascade and cell apoptosis, a process now described as secondary axotomy.
Conclusion
The difference in TAI definitions makes the comparison of studies that report outcomes, treatments, and prognostic factors a daunting task. An even more difficult task is isolating the outcomes of isolated TAI from the outcomes of severe TBI in general. Targeted bench-to-bedside studies are required in order to uncover further pathways involved in the pathophysiology of TAI and, ideally, new treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic axonal injury (TAI) is a condition characterize as multiple, scattered, small hemorrhagic, and/or non-hemorrhagic lesions, alongside brain swelling, in a more confined white matter distribution on imaging studies, together with impaired axoplasmic transport, axonal swelling, and disconnection with more than 3 such foci present on imaging studies according to the National Institutes of Health Common Data Elements [1]. In the past, DAI (diffuse axonal injury) was defined as prolonged (> 6 h) loss of consciousness (LOC), without a visible mass lesion.
TAI was first described mid-twentieth century, as diffuse microscopic pathological changes to the brain tissue [2]. These lesions were believed to be the direct result of mechanical impact on brain tissue after trauma. This belief later relied on performing experimental observations using gelatine models of the human brain, where shear and tear was found after applying rotational force of relative intensity to those in a traumatic incident [3, 4].
At the start of the 1980s, the term DAI (diffuse axonal idiopathic injury, nowadays called TAI) was introduced in consecutive studies by Adams and Gennarelli, among others [5, 6]. The term DAI implicates that there is diffuse topographic distribution of traumatic findings. However, studies showed that the distribution of traumatic lesions in DAI has a predisposition for white matter tracts in the midline of the brain, including the corpus callosum, internal capsule, cerebral peduncles, brainstem, and the grey-white junction of the cerebral cortex [5, 7]. These findings are probably related to the type of post-mortem examination done in the study with higher prevalence of central traumatic lesions after high energy trauma. Nowadays, the term TAI, or traumatic axonal injury, is more accurate than the term DAI.
TAI is thought to be caused by a variety of traumatic mechanisms involving fast acceleration and/or deceleration, including motor vehicle accidents, falls from height, and blunt assault [6]. Adams et al. suggested a grading system for TAI, dividing TAI into three different subgroups (Table 1) [5]. In clinical practice, this grading has been applied clinically using MRI susceptibility-weighted (SW) imaging, but no thorough external validation studies have been carried out to confirm proper clinical use of the MRI grading.
Materials and methods
We performed a search from the first available records of each specific database to July 2020 using MEDLINE, Embase, Web of Science, and Google Scholar with the terms “diffuse axonal injury,” “traumatic axonal injury,” and “axonal injury.” The references of all included articles were also cross-checked for missed articles. Both clinical and basic science papers were included, describing the pathophysiology, diagnostic studies, and TAI-targeted treatments. Both pediatric and adult studies were included.
Two independent reviewers sifted through the title/abstracts and decided on the full-text inclusions. These were reviewed in the second sift phase and conflicts were resolved by consulting a third reviewer. A total of 160 articles were included.
We designed a scoping, narrative review with the objective to synthetize available information and identify the knowledge hiatus in the literature. A preliminary orientating search showed great heterogeneity in study methods and research type. Therefore, we did not expect enough comparable data and we also did not expect it to be of sufficient quality in order to perform a meta-analysis, so this was not defined in our study protocol. Therefore, we did not publish a protocol on Prospero, we did not plan any pooled analysis or sensitivity analysis, but we did perform a risk of bias analysis for most included papers.
Pathophysiology
The pathophysiology of TAI is complex and lacking a unifying theory. The assumption that TAI is primarily and solely caused by direct mechanical force has been abandoned. Besides the primary damage, there is secondary damage caused by chemical alterations and changes in neuronal metabolism.
Trauma mechanism
There are two major mechanisms involved in head trauma: direct impact and accelerative and decelerative (a/d) forces [8]. Sudden head movement produces a force vector inside the intracranial cavity, resulting in shearing and strain injury. Shear and tear of axonal fibers can cause axonal damage, resulting in TAI [6].
The duration of the a/d forces is decisive for the type of injury. Slower A/d forces with a relatively long duration (20–25 ms) will mainly cause TAI, whereas shorter duration of a/d forces will likely cause acute subdural hematoma (ASDH) through shearing of bridging veins [6, 9]. A/d forces in the coronal plane are primarily associated with the occurrence of TAI [6, 10, 11]. The combination of translational and angular acceleration [12], but also rotational acceleration [13] has also been suggested as the most prominent cause of TAI.
The fact that TAI can be caused by traumas with relatively low rates of acceleration is of importance in forensic pathology. Deadly TAI can occur even if the initial impact force is not strong enough to cause fracturing of the skull or evident macroscopic pathology to the brain [10, 14].
Primary axotomy
The initial hypothesized pathophysiological mechanism was that pure mechanical stretch due to traumatic acceleration or deceleration alone led to direct tearing of axonal fibers. Subsequently to this tearing, the damaged axonal fibers would retract and form retraction bulbs, which are visible on pathological examination. This process of direct tearing is called primary axotomy [15, 16]. Gliding hemorrhages visible on neuroimaging right after trauma suggest that primary axotomy can occur directly after impact. Complete primary axotomy is nowadays considered a rare form of damage that occurs in cases of massive, widespread axonal damage [8, 17, 18]. Primary axotomy may also cause a decreased level of consciousness through widespread CNS damage including the thalami and the reticular substance of the brainstem.
Secondary axotomy: molecular basis
If the inertial forces are of low intensity and do not cause complete primary axotomy, they can still be strong enough to cause partial damage to the axon, triggering a molecular pathway resulting in what is nowadays called secondary axotomy [19, 20]. Secondary axotomy is an inflammatory and apoptotic event (Fig. 1), which may create a potential window of opportunity for therapeutic treatment [21]. This window will probably only be a few hours. While the primary axotomy is a true mechanical event, caused by shearing forces, secondary axotomy can be seen as an apoptotic/neurodegenerative event which result in secondary loss of axons after trauma. Secondary axotomy can also be seen as a continuation of primary axotomy, where the initial structural damage caused by the traumatic forces forms the base for the entire molecular cascade, instead of as a separate entity.

Primary and secondary axotomy pathophysiological processes (left-hand inlay), courtesy of M. W. T. van Bilsen, MD. The figure illustrates an axon upon which shear and rotational forces act. The microtubules (blue) become progressively stiffer and eventually break, leading to a disruption of the axonal transport of molecules. Calcium accumulates in the cell, both through the mechanical opening of calcium channels, as well as through the disruption of mitochondria. Through hydrolyzation of calpastatin (which normally inhibits calpain), calpain is activated and it in turn hydrolyzes the cytoskeleton and microtubules. This cascade leads to apoptosis and axon disconnection
The key element in secondary axotomy is axonal microtubules (MT). MT are subcellular structural cytoskeletal elements composed of long, stiff polymers of αβ-tubulin. MT are structurally and functionally important components of the eukaryotic cell cytoskeleton, which can serve as tracks for intracellular trafficking and energy-dependent directional intracellular transport [22]. MT are viscoelastic. With rapid stretch, the MT become the stiffest portion of the axon, making them viable to breaking (Fig. 1). MT breakage interrupts the axonal transport, leading to accumulation of transport proteins such as β-APP. The accumulation of these proteins causes axonal swelling, which can lead to disconnection of the axon from the neuron body if the process evolves severely enough. The primary mechanical breakage of MT can be followed by secondary depolymerisation of the remaining MT, disrupting axonal transport even more [23,24,25,26]. Mechanical damage to axonal microtubules itself can also be considered as incomplete primary axotomy.
Another major player in the secondary axotomy is the increased concentration of intracellular calcium ions [27]. Multiple mechanisms lead to increase in intracellular calcium, including activity of calcium-ATPase, the release of intra-axonal calcium storage, and the mechanical dysregulation of voltage-gated sodium channels [28,29,30,31].
The increased intracellular calcium concentration causes multiple intracellular processes leading to cell death. The cysteine and caspase-3 protein pathways become activated [32]. Hydrolyzation of calpastatin, a calpain inhibitor, leads to accumulation of calpain, which subsequently hydrolyses multiple structural and functional proteins within 1 to several hours after injury [27, 33,34,35,36]. Calpain-induced hydrolyzation of the sodium channel inactivation gate leads to progressively increasing intracellular calcium concentrations and calcium-associated damage [37]. Hydrolyzation of spectrin leads to severe damage to the cytoskeletal network. Microtubules and neurofilaments also get hydrolysed and thereby damaged. As a result of cytoskeletal degradation, axons get instable and disconnect [27, 38,39,40,41,42,43,44,45,46,47,48,49,50].
Calpain also has a role in ankyrin proteolysis. This may lead to distortion of the arrangement of sodium channels on the nodes of Ranvier. This pathway could contribute to instability of the axonal membrane [51].
Increased intracellular levels of calcium alter the permeability of mitochondrial membrane through the activation of calcineurin [52]. Activation of oxygen radicals also contributes to changes in mitochondrial membrane permeability. Calcium overload further causes swelling and rupture of the mitochondria. This influences homeostasis, energy metabolisms, and the release of caspases. Caspase hydrolyses proteins in severely damaged axons, resulting in caspase-mediated programmed cell death [53,54,55,56]. Mitochondrial failure leads to the inability to produce high energy phosphates, which are needed for the support of axonal functions and structure [56].
This concerted action of MT stiffness on the one hand and calcium overload with apoptosis and cell death are what ultimately lead to neuron dysfunction and widespread loss of connectivity. Secondary axotomy is nowadays accepted as the mechanism of neuronal dysfunction after TAI but the exact timing of events and the targeted treatment remain elusive.
Currently, the role played by myelin and glial cells in the process of secondary axotomy constitutes an important focus of research. In the central nervous system, wrapping of oligodendrocytes plasma membrane extensions forms the myelin sheath around the axons [57]. Myelin is thought to play a role in maintaining axon stability and integrity [58,59,60]. Animal studies reveal that there is significant loss of myelin in the corpus callosum and the brain stem after TAI. The vulnerability of oligodendrocytes and the demyelination that follows after oligodendrocyte apoptosis may contribute to axonal damage in the brainstem. Myelin and oligodendrocytes, as well as their interplay, might form a future therapeutic target for the treatment of TAI [61].
TAI, coma, and loss of consciousness
In the past, DAI was defined (due to a lack of more advanced imaging studies and limited understanding of pathophysiological mechanisms) by prolonged loss of consciousness (LOC) (> 6 h), in the absence of a visible mass lesion [62, 63]. Recently, isolated TAI as the sole etiology of coma has been called into question [64, 65].
Biomechanically, the plane of the brainstem relative to the rotational force might be involved in the occurrence and duration of LOC. Whereas rotation of the brainstem on its transverse axis was associated with coma, equal rotation on the vertical axis was not [56, 66]. LOC occurs thus even when small rotational forces act transversally to the brainstem [67].
The topographic distribution of axonal damage is also important for the occurrence of LOC [56]. Duration of coma has been linked directly to the severity of TAI in the brainstem, but more extensive bilateral thalamic damage or the massive, diffuse hemispheric damage likely also play a role [66]. Despite having an essential role, the brain stem is often not the only cause of coma in TAI patients.
Diagnostics
Histopathological findings
Histopathology only plays a role in post-mortem diagnostics. It is not used in a clinical setting. The traditional histological findings in TAI are large axonal dilations caused by complete axotomy, referred to as retraction bulbs or axonal bulbs [2, 5, 23, 68,69,70,71,72,73,74,75]. Axonal varicosities are another histological finding. These are dilations along the length of the axon, visible within several hours after trauma, the result of processes described above under secondary axotomy due to axonal transport impairment and protein accumulation [19, 25, 26, 71].
Traditionally, hematoxylin and eosin (HE), and several silver stains were most widely used to detectpathological changes. Nowadays, immunohistochemical staining is more widely used. Accumulation of beta-amyloid precursor protein (β-APP) is a sensitive marker for diagnosis of TAI [76,77,78,79,80]. Accumulation of β-APP is visible within 2 h after trauma and shows more extensive injury than HE or silver staining. Because of these issues, β-APP immunostaining is the golden standard for pathological diagnosis of TAI [81,82,83]. It must be stated that the characteristic histological observations found appear after distinct times post-trauma and depend on the survival times of the individual affected (before the brain was harvested for pathological analysis).
Imaging studies
CT is capable of identifying large TAI-related hemorrhage, but non-hemorrhagic lesions and small TAI hemorrhage are virtually impossible toidentify using CT [84]. The slice thickness of a conventional trauma head CT is about 5–10 mm. Since TAI lesions may fall under this detection margin, they can be easily missed using conventional CT.
Conventional MRI (cMRI) has a higher sensitivity in demonstrating lesions in the brainstem and the deep white matter, making it more sensitive for identifying axonal injury compared to CT [85,86,87,88].
The MRI gradient echo sequence (GRE) is able to detect heme and heme breakdown products, making it a suitable method for discovering small hemorrhagic lesions [89].
Susceptibility weighted imaging (SWI) as a variant sequence of GRE imaging should be considered the “gold standard” for identifying TAI lesions. It has a higher sensitivity for hemorrhage than GRE , which makes it more useful for early diagnosis of TAI [84, 90]. However, association with outcomes has not yet been demonstrated and SWI might overestimate the size of a lesion due to its high sensitivity to heme products (Fig. 3).
Diffusion-weighted imaging (DWI) can accurately examine non-hemorrhagic lesions [84]. High signal DWI can be used in patients with early stage TAI [91]. Lesions found represent cellular swelling and cytotoxic oedema [92]. DWI may aid in predicting clinical outcome after TAI [93]. DWI is more capable of determining the severity of the injury and estimating the long-term prognosis than MRI techniques [94].
Diffusion tensor imaging (DTI) is an improved form of DWI. It can be used to evaluate nerve alignment, white matter microstructure and the morphology around nerve fibers [84]. Within the first 24 h after trauma, DTI can detect white matter regions with reduced anisotropy, making it an adequate technique for detecting TAI [95]. Anomalies found with DTI seem to be associated with both acute GCS and modified Rankin scores (mRS) at discharge [96]. Animal studies comparing DTI images and histological examination showed that DTI is highly sensitive for axonal injury and likely has a high negative predictive value [97, 98].
The direct correlation of histopathologic findings with MRI findings has not been validated in large human studies, which makes the interpretation of the TAI grade as proposed by Adams and colleagues [5] impossible based on imaging studies. Caution needs to be used when employing this tactic in a clinical setting, despite the practice being widespread.
The longer scanning time and inability to closely monitor patients undergoing MRI makes this technique less relevant in the acute phase after trauma, save for recent research endeavors. Many patients are not stabile enough to withstand the long scanning time used to make MRI images. CT-scanning is more widely used in the acute phase, due to its shorter scanning time. MRI scanning should be performed as soon as the condition of the patient allows it, so the full extent of trauma can be mapped and white matter volume prospectively followed-up.
Biochemical markers
Several potential biochemical markers have been investigated in TAI. Most of these are being investigated in the broader sense of their evolution post-severe TBI. C-tau, the polymerized version of the microtubule-related protein Tau, might be one of the most useful biomarkers for diagnosing TAI [99]. The detection level of C-tau in the cerebrospinal fluid (CSF) appears to be related to the severity of TAI in the clinical setting [45, 100]. The protein tau on itself is also a potential biomarker. Significantly higher serum levels of tau have been found in patients with TAI compared to patients without TAI; however, tau levels were not a predictor for unfavorable outcome in TAI patients [101].
Neurofilamental heavy chains (NF-H) are an emerging marker for diagnosing TAI, together with light chains (NF-L), which in turn appear to be a highly sensitive and specific marker for TAI [102, 103].
The biomarkers S-100B and neuron-specific enolase (NSE) are also used both in research and clinically. Both can be detected at increased levels in serum and CSF after TBI [104,105,106,107,108,109,110,111,112,113,114]. The serum biomarker levels seem to be associated with the cumulative size and number or contusion detected by CT-imaging [115]. S-100B correlates more with the severity of TBI [116]. Normal levels of S-100B after trauma might accurately exclude brain injury, as is the practice in northern countries [117].
Besides being the histological golden standard, β-APP is also of interest as a biochemical marker. Very basic research into β-APP derivatives in blood and in cerebrospinal fluid leads to the suggestion that 𝛽-APP derivatives in CSF could be an indicator of axonal damage [118].
Plasma levels of high-density lipoprotein cholesterol (HDL-C) are an emerging diagnostic biomarker to diagnose for TAI. In a retrospective study, HDL-C levels within 1 week after trauma of TAI patients were significantly lower than in the non-TAI control group. HDL-C was identified as an independent predictor for TAI after multivariable analysis [119].
Several studies in the field of proteomics have been conducted, generating long lists of potential biomarker proteins [120, 121]. Proteomics might be a promising field of research to find suitable biomarkers in the future.
A wealth of biomarker research is being conducted at this moment. Through better characterization of the disease phenotype, hope exists for potential future personalized treatment.
Secondary axotomy-targeted treatment
Treatment of TAI mainly consists of treating underlying problems, the focal and systemic injuries. Currently, there is no TAI-specific treatment available. Treatment is performed in comformity with the “Guidelines for the Management of Severe Traumatic Brain Injury” [122].
However, a lot of research aims to identify possible treatment options based on the molecular theory explained above. Two main treatment types can be identified. The first type are treatments that focus on the prevention of secondary axotomy. Since secondary axotomy is an inflammatory and apoptotic event (Fig. 2), there may be a potential window of opportunity for therapeutic treatment [21]. The second type aims to enhancing neuronal regeneration after damage has already occurred.

Reproduced with permission. YFP+ axonal swellings progressively enlarge and disconnect over time post-injury. Consistent with progressive axonal change, YFP+ swellings continue to enlarge over the next 12 h post-injury (a–c). Though the majority of swellings progress to disconnection over this 12-h time span, occasional swellings still maintaining axonal continuity could be found at later time points following injury (d–g), underscoring a temporally heterogeneous evolution of YFP+ swellings over time. Scale 10 μm. Reprinted by permission from Springer Nature: Acta Neuropathol Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment, Greer et al, Copyright 2013
Treatment preventing secondary axotomy
Multiple agents have been studied that influence calcium hemostasis. Administration of the Ca-channel blocker nimodipine could play a role in preventing or minimizing secondary damage of the axon. Preclinical studies showed a decreased expression of 𝛽-APP, suppressed activity of calcineurin (CaN), and lessened ultrastructural axonal damage [52]. Ciclosporin A (CsA) also inhibits CaN. By preventing a rise in mitchondrial membrane permeability, CsA abates swelling and disruption of mitochondria and thereby mitigates axonal damage [27, 38, 123, 124]. It also antagonizes calcium-mediated cytoskeletal disruption, and thereby secondary axotomy [27, 38, 125].
The calpain inhibitors MDL28170, 5b, AK295, and SJA-6017 seem to have a protective value; however, this protective effect has been disputed [126,127,128,129,130].
FK506 (also known as tacrolimus) seems to be less toxic than CsA [131]. It attenuates CaN, without a mitochondrial effect, even though this has been disputed in recent studies. Despite this, FK506 provides massive axonal protection and decreased axonal injury [132,133,134]. By reducing progressive damage to the cytoskeleton, FK506 also reduces secondary axotomy [131].
In a more recent study, the drugs SN-6 and dynasore have been proven to both reduce the extend of stretch injury-induced swelling of axonal varicosities and mitochondrial degradation. It also seems to protect against oxidative damage. SN-6 inhibits the the sodium calcium exchanger1 (NCX1), whereas dynasore inhibits several GTPase proteins (Drp1, dynamin1 and 2) which regulate mitochondrial cleavage [135].
Microtubules form another target for therapy. Taxol-like drugs (taxanes) are microtubule stabilizing drugs. Paclitaxel (Taxol) has possible effect on limiting axonal degeneration [26, 136, 137]. In vitro paclitaxel effectuated secondary depolymerization of microtubules, reducing axonal swelling and degeneration [26]. Since Paclitaxel cannot easily pass the blood-brain barrier, high IV doses are required leading to severe side-effects, such as peripheral neuropathy [138]. The microtubule stabilizing drug Epothilone D is more capable of passing the blood-brain barrier. It is shown to be beneficial in spinal cord injury, but there is no proof of its benefit in brain injury [139, 140].
Combination of hypothermia with drugs like FK506 or CsA during the rewarming phase might give additional protection [134, 141, 142]. This is either caused by a synergistic effect or because hypothermia reduces the progression of TAI, prolonging the window of opportunity for drug treatment [142,143,144,145].
Erythropoietin (EPO) has been shown improve both motor deficit and cognitive functions in an animal model, and was able to preserve axons in the injured area [146]. However, EPO can increase the calcium influx [147]. This can promote TAI progression in the early phase, so its applicability remains limited [148]. A recent RCT showed no improvement of neurological outcome 6 months after TBI after administration of erythropoietin [149, 150].
The neuroprotective properties of progesterone have also been studied. Results are conflicting. Studies show that progesterone might yield better learning, memory, cognitive, and motor functions and reduced axonal damage [151, 152], and improved neurological outcome on the long term [153]. However, a large meta-analysis showed no effect of progesterone in reducing mortality or unfavorable outcomes [154].
A recent study found altered expression of α7nAChR, a nicotine receptor, in a rat TAI model. They found that administration of a selective α7nAChR agonist yielded better learning and memory performance as compared to a α7nAChR antagonist [155].
Treatment enhancing neuronal regeneration
Stem cell therapy is studied not only in the field of TBI, but also in the field of ischemic brain damage. The therapeutic value of stem cells is traditionally believed to be based on two principles. There is thought to be direct cell replacement by stem cells. On the other hand, stem cells are thought to release neurotrophic factors, enhancing neuronal regeneration. A newer idea is that of bio-bridging. The idea behind this theory is that the stem cells form a connection between the areas on both sides of the neuronal damage. This so-called bio-bridge can form a pathway that promotes both proliferation and migration of native stem cells [156].
In the context of TAI, implantation of embryonal stem cells (ESC), neuronal derived stem cells (NSC), and bone-marrow-derived stem cells (BMSC) has been researched. Implantation of embryonal stem cells (ESC) after TAI may yield improvement in functional outcome [157]. The tumorigenic potential of ESC seems to be reduced when cell is pre-differentiated in vitro [158, 159].
Neural stem cells (NSC) can improve motoric recovery and cognition [160,161,162].
Bone-marrow-derived stem cells (BMSC) showed a significantly improved functional outcome (as measured by the results of the rotarod test and the modified neurological severity score) in rats [161]. BMSC have been proven migrate to the injured brain and to improve cognitive dysfunction related to TAI, when transplanted 10 days after trauma [163]. BMSC survived after both intra-arterial and intravenous administration, after which cells migrated to the site of injury expressing neural cell markers [164, 165].
It is not entirely clear whether the beneficial effect of stem cell transplant is caused by either direct cell replacement, neurotrophic factors of bio-bridging. It has been shown that neurotrophic factors are released after stem cell transplant, leading to the conclusion that they form a key role in neuronal regeneration after stem cell implant [157, 160, 162].
Stem cell therapy has only been tested in vitro and in animal studies. No inpatient studies with stem cells have been performed so far. Since stem cell therapy focuses on neuronal regeneration and repair, it is probably more valuable in the chronic reparative phase and not in the acute stage of ICU management. Solid evidence on the exact timing and effectiveness of stem cell therapy is not yet available.
Neuronal axons of the central nervous system lack regenerative capacities, caused by a variety of pathways. Multiple agents addressing these pathways have been investigated, showing possible improvement of axonal outgrow and functional recovery in preclinical studies [166, 167].
Other studies focus on components the axonal membrane and support membrane repair [43, 168,169,170].
Despite a wealth of research into targeted treatments for TAI, a more bench-to-bedside approach is needed before we expect to have a breakthrough in this respect. A possible explanation for the lack of human treatment, despite the wealth of animal studies, is the incongruence in the pathological timelines between humans and rodents. There appears to be no suitable conversion rate between the two [171]. This makes the translation from experimental studies into clinical practice difficult.
Most important findings
In order to make a clear distinction between diffuse axonal injury by non-traumatic causes and diffuse axonal injury due to trauma, we suggest that the term traumatic axonal injury, or TAI, should be used. The term DAI should be abandoned when speaking purely about the traumatic version.
Direct shear injury and tearing of axonal fibers seems to be of less clinical importance than the secondary axotomy, caused by a cascade of molecular pathways. Primary damage is a result of more massive injury and therefore probably does not exist in all TAI patients. Besides this, primary damage already occurs prior to arrival at the hospital and therefore leaves little to no options for treatment. Secondary axotomy is a slower process, and therefore leaves a window of opportunity for treatment.
Histopathological assessment of TAI injury has more historic rather than clinical relevance. Imaging, however, is of great clinical importance. MRI, and various on MRI-based techniques, seem to have the highest capability of identifying TAI lesions (Fig. 3). However, logistic concerns and safety during transportation to and from the ICU makes most polytrauma and severe TBI-patients are only eligible for CT-scanning. Evolution of MRI techniques will likely provide a breakthrough in this respect in the future.

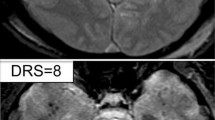
Susceptibility-weighted imaging (SWI) MRI series of a young patient with TAI. Note the susceptibility artifacts in the brain stem, temporal lobes, and cerebellum (a) and splenium of the corpus callosum (b). Despite grade 3 TAI, the patient was awake and communicating
Ideally, there should be a biomarker to identify TAI and to predict outcome. Currently, there are several potential biomarkers being researched. However, most of these biomarkers seem to be nonspecific for TAI, since they are present in all types of severe TBI. A biomarker specific for TAI has yet to be identified, but is a highly important area of future research, including research in the field of proteomics.
Currently, treatment enhancing regeneration of neural cells is not available. Some fundamental in vitro research on stem cell therapy seems promising in experimental studies. No studies have shown effectiveness of this therapy yet. The potential tumorigenic potential of stem cells calls for great caution with this technique into practice [158, 159]. Since the effect of stem cell transplantation might be caused by neurotropic factors, administration of these factors alone might be a promising field of research.
Treatment preventing secondary axotomy seems to be most promising. Avoiding axonal damage is better than repairing it. A plethora of experimental research is directed at finding a possible treatment in the window of opportunity between the primary and secondary axotomy phases. A bench-to-bedside approach from centers with broad experience in the treatment of TBI is required.
In daily practice, a cumbersome factor in both research and clinical decision-making for patients with TAI is the unpredictability of the outcome. Since many of the clinical decisions and informed consent talks are based on outcome data from the scientific literature, it is essential to commit further scientific efforts to report long-term longitudinal data of TAI patients, both isolated and within large severe TBI cohorts. Evidence in TBI, despite massive research efforts, remains weak [172]. Together with fundamental research efforts, this will bring new insights into the potential treatment optimization and future possibilities for TAI patients.
Clinical highlights and key points
-
Primary axotomy is a mechanical lesion of the axon which occurs at the moment of trauma, followed by a “secondary” axotomy, involving apoptotic and degenerative processes (hydroxylation of proteins), especially at the level of the microtubules.
-
Both primary and secondary axotomy may be the cause of coma despite the absence of increased intracranial pressure, depending on the foci of injury (e.g., bilateral thalamic and brainstem reticular formation).
-
The Adams classification has never been validated in MRI studies. “Grade III” TAI, seen on imaging studies and sometimes used as a marker of the severity of the lesions, has no proven association with outcomes.
-
TAI should be treated the same way as severe TBI, according to the guidelines, but centers with interest in TBI research should be aware of bench-to-bedside research efforts and participate in clinical trials for promising compounds from basic science research.
-
As with other investigated therapies, timing is essential in TBI. The window of opportunity between primary and secondary axotomy is short.
Conclusion
The prognostic value of TAI lesions seen on susceptibility-weighted MRI series has not yet been elucidated. Whether TAI requires targeted treatment different from regular severe TBI ICP-targeted therapy is still unclear. The chain of events involved in the pathophysiology further research in order to improve patient outcomes and potentially discover targeted therapies.
References
NINDS Common Data Elements (2012) Traumatic brain injury: data standards. https://www.commondataelements.ninds.nih.gov/Traumatic Brain Injury. Accessed 12-09-2020
Rand CW, Courville CB (1946) Histologic changes in the brain in cases of fatal injury to the head; alterations in nerve cells. Arch Neurol Psychiatr 55:79–110
Holbourn AHS (1943) Mechanics of head injury. Lancet 242:438–441
Holbourn AHS (1945) Mechanics of brain injuries. Br Med Bull 3:147–149
Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR (1989) Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 15:49–59
Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP (1982) Diffuse axonal injury and traumatic coma in the primate. Ann Neurol 12:564–574
Adams JH, Graham DI, Gennarelli TA, Maxwell WL (1991) Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry 54:481–483
Gennarelli TA (1993) Mechanisms of brain injury. J Emerg Med 11(Suppl 1):5–11
Oehmichen M, Meissner C, Schmidt V, Pedal I, König HG, Saternus KD (1998) Axonal injury—a diagnostic tool in forensic neuropathology? A review. Forensic Sci Int 95:67–83
Yoganandan N, Gennarelli TA, Zhang J, Pintar FA, Takhounts E, Ridella SA (2009) Association of contact loading in diffuse axonal injuries from motor vehicle crashes. J Trauma 66:309–315
Zhang J, Yoganandan N, Pintar FA, Gennarelli TA (2006) Role of translational and rotational accelerations on brain strain in lateral head impact. Biomed Sci Instrum 42:501–506
Wang HC, Duan ZX, Wu FF, Xie L, Zhang H, Ma YB (2010) A new rat model for diffuse axonal injury using a combination of linear acceleration and angular acceleration. J Neurotrauma 27:707–719
Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA (1995) Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma 12:689–694
Yoganandan N, Baisden JL, Maiman DJ, Gennarelli TA, Guan Y, Pintar FA, Laud P, Ridella SA (2010) Severe-to-fatal head injuries in motor vehicle impacts. Accid Anal Prev 42:1370–1378
Adams H, Mitchell DE, Graham DI, Doyle D (1977) Diffuse brain damage of immediate impact type. Its relationship to 'primary brain-stem damage' in head injury. Brain 100:489–502
Stirch SJ (1956) Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry 19:163–185
Geddes JF, Vowles GH, Beer TW, Ellison DW (1997) The diagnosis of diffuse axonal injury: implications for forensic practice. Neuropathol Appl Neurobiol 23:339–347
Menon DK, Schwab K, Wright DW, Maas AI (2010) Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil 91:1637–1640
Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT (1994) Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma 11:173–186
Greer JE, Hånell A, McGinn MJ, Povlishock JT (2013) Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathol 126(1):5–74. https://doi.org/10.1007/s00401-013-1119-4
Povlishock JT, Jenkins LW (1995) Are the pathobiological changes evoked by traumatic brain injury immediate and irreversible? Brain Pathol 5:415–426
Xu K, Zhong G, Zhuang X (2013) Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339:452–456
Povlishock JT (1992) Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 2:1–12
Povlishock JT, Christman CW (1995) The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma 12:555–564
Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH (2012) Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol 233:364–372
Tang-Schomer MD, Patel AR, Baas PW, Smith DH (2010) Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J 24:1401–1410
Büki A, Povlishock JT (2006) All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir 148:181–193
Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH (2004) Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci 24:4605–4613
Maxwell WL, McCreath BJ, Graham DI, Gennarelli TA (1995) Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J Neurocytol 24:925–942
Staal JA, Dickson TC, Gasperini R, Liu Y, Foa L, Vickers JC (2010) Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J Neurochem 112:1147–1155
Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci 21:1923–1930
Büki A, Okonkwo DO, Wang KK, Povlishock JT (2000) Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci 20:2825–2834
Donald CLM, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, Snyder AZ, Raichle ME, Witherow JR, Fang R, Flaherty SF, Brody DL (2011) Detection of blast-related traumatic brain injury in U.S. military personnel. N Engl J Med 364:2091–2100
Huang L, Obenaus A (2011) Hyperbaric oxygen therapy for traumatic brain injury. Med Gas Res 1:21
McGowan JC, Yang JH, Plotkin RC, Grossman RI, Umile EM, Cecil KM, Bagley LJ (2000) Magnetization transfer imaging in the detection of injury associated with mild head trauma. AJNR Am J Neuroradiol 21:875–880
Sotelo JR, Canclini L, Kun A, Sotelo-Silveira JR, Calliari A, Cal K, Bresque M, Dipaolo A, Farias J, Mercer JA (2014) Glia to axon RNA transfer. Dev Neurobiol 74:292–302
von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH, Siman R, Meaney DF (2009) Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit. J Neurosci 29:10350–10356
Büki A, Okonkwo DO, Povlishock JT (1999) Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma 16:511–521
Büki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol 58:365–375
Farkas O, Polgár B, Szekeres-Barthó J, Dóczi T, Povlishock JT, Büki A (2005) Spectrin breakdown products in the cerebrospinal fluid in severe head injury—preliminary observations. Acta Neurochir 147:855–861
Jeanneret V, Yepes M (2016) The plasminogen activation system promotes dendritic spine recovery and improvement in neurological function after an ischemic stroke. Transl Stroke Res. https://doi.org/10.1007/s12975-016-0454-x.
Kampfl A, Posmantur R, Nixon R, Grynspan F, Zhao X, Liu SJ, Newcomb JK, Clifton GL, Hayes RL (1996) mu-calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem 67:1575–1583
Kilinc D, Gallo G, Barbee K (2007) Poloxamer 188 reduces axonal beading following mechanical trauma to cultured neurons. Annu Int Conf IEEE Eng Med Biol Soc 2007:5395–8. https://doi.org/10.1109/IEMBS.2007.4353562
Kilinc D, Gallo G, Barbee KA (2008) Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp Neurol 212:422–430
Kilinc D, Gallo G, Barbee KA (2009) Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp Neurol 219:553–561
McGinn MJ, Kelley BJ, Akinyi L, Oli MW, Liu MC, Hayes RL, Wang KK, Povlishock JT (2009) Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J Neuropathol Exp Neurol 68:241–249
Merali Z, Leung J, Mikulis D, Silver F, Kassner A (2015) Longitudinal assessment of imatinib's effect on the blood-brain barrier after ischemia/reperfusion injury with permeability MRI. Transl Stroke Res 6:39–49
Mondello S, Robicsek SA, Gabrielli A, Brophy GM, Papa L, Tepas J, Robertson C, Buki A, Scharf D, Jixiang M, Akinyi L, Muller U, Wang KK, Hayes RL (2010) αII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. J Neurotrauma 27:1203–1213
Mustafa AG, Wang JA, Carrico KM, Hall ED (2011) Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem 117:579–588
Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK (1996) Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol 55:850–860
Reeves RM, Greer JE, Vanderveer AS, Phillips LL (2010) Proteolysis of submembrane cytoskeletal proteins ankyrin-G and αII-spectrin following diffuse brain injury: a role in white matter vulnerability at Nodes of Ranvier. Brain Pathol 20:1055–1068
Mu J, Song Y, Zhang J, Lin W, Dong H (2015) Calcium signaling is implicated in the diffuse axonal injury of brain stem. Int J Clin Exp Pathol 8:4388–4397
Badaut J, Bix GJ (2014) Vascular neural network phenotypic transformation after traumatic injury: potential role in long-term sequelae. Transl Stroke Res 5:394–406
Friess SH, Ichord RN, Owens K, Ralston J, Rizol R, Overall KL, Smith C, Helfaer MA, Margulies SS (2007) Neurobehavioral functional deficits following closed head injury in the neonatal pig. Exp Neurol 204:234–243
Tate CM, Wang KK, Eonta S, Zhang Y, Carr W, Tortella FC, Hayes RL, Kamimori GH (2013) Serum brain biomarker level, neurocognitive performance, and self-reported symptom changes in soldiers repeatedly exposed to low-level blast: a breacher pilot study. J Neurotrauma 30:1620–1630
Xiao-Sheng H, Sheng-Yu Y, Xiang Z, Zhou F, Jian-ning Z (2000) Diffuse axonal injury due to lateral head rotation in a rat model. J Neurosurg 93:626–633
Simons M, Trotter J (2007) Wrapping it up: the cell biology of myelination. Curr Opin Neurobiol 17:533–540
Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA (1998) Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280:1610–1613
Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374
Yin X, Baek RC, Kirschner DA, Peterson A, Fujii Y, Nave KA, Macklin WB, Trapp BD (2006) Evolution of a neuroprotective function of central nervous system myelina. J Cell Biol 172:469–478
Mu JLM, Wang T, Li X, Bai M, Zhang G, Kong J (2019) Myelin damage in diffuse axonal injury. Front Neurosci 13:217
Alberico AM, Ward JD, Choi SC, Marmarou A, Young HF (1987) Outcome after severe head injury. Relationship to mass lesions, diffuse injury, and ICP course in pediatric and adult patients. J Neurosurg 67:648
Eum SW, Lim DJ, Kim BR, Cho TH, Park JY, Suh JK, Lee KC, Lee HK (1998) Prognostic factors in patients with diffuse axonal injury. J Korean Neurosurg Soc 27:1998–1674
Firsching R, Woischneck D, Klein S, Reissberg S, Döhring W, Peters B (2001) Classification of severe head injury based on magnetic resonance imaging. Acta Neurochir 143:263–271
Rosenblum WI (2015) Immediate, irreversible, posttraumatic coma: a review indicating that bilateral brainstem injury rather than widespread hemispheric damage is essential for its production. J Neuropathol Exp Neurol 74:198–202
Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF (2000) Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg 93:315–322
Browne KD, Chen XH, Meaney DF, Smith DH (2011) Mild traumatic brain injury and diffuse axonal injury in swine. J Neurotrauma 28:1747–1755
Adams JH, Doyle D, Graham DI, Lawrence AE, McLellan DR (1984) Diffuse axonal injury in head injuries caused by a fall. Lancet 2:1420–1422
Adams JH, Graham DI, Murray LS, Scott G (1982) Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol 12:557–563
Chen X-H, Meaney DF, Xu B-N, Nonaka M, McIntosh TK, Wolf JA, Saatman KE, Smith DH (1999) Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol 58:588–596
Povlishock JT, Becker DP (1985) Fate of reactive axonal swellings induced by head injury. Lab Investig 52:540–552
Povlishock JT, Becker DP, Cheng CLY, Vaughan GW (1983) Axonal change in minor head injury. J Neuropathol Exp Neurol 42:255–242
Povlishock JT, Buki A, Koiziumi H, Stone JJ, Okonkwo DO (1999) Initiating mechanisms involved in the pathobiology of traumatically induced axonal injury and interventions targeted at blunting their progression. Acta Neurochir Suppl 73:15–20
Povlishock JT, Katz DI (2005) Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil 20:76–94
Smith DH, Meaney DF (2000) Axonal damage in traumatic brain injury. Neuroscientist 6:483–495
Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW (1993) Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett 160:139–144
Grady MS, McLaughlin MR, Christman CW, Valadka AB, Fligner CL, Povlishock JT (1993) The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol 52:143–152
Gultekin SH, Smith TW (1994) Diffuse axonal injury in craniocerebral trauma. A comparative histologic and immunohistochemical study. Arch Pathol Lab Med 118:168–171
Ng HK, Mahaliyana RD, Poon WS (1994) The pathological spectrum of diffuse axonal injury in blunt head trauma: assessment with axon and myelin strains. Clin Neurol Neurosurg 96:24–31
Sherriff FE, Bridges LR, Sivaloganathan S (1994) Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol 87:55–62
Gentleman SM, Roberts GW, Gennarelli TA, Maxwell WL, Adams JH, Kerr S, Graham DI (1995) Axonal injury: a universal consequence of fatal closed head injury?. Acta Neuropathol 89(6):537–43. https://doi.org/10.1007/BF00571509
Lee SR, Choi B, Paul S, Seo JH, Back DB, Han JS, Choi DH, Kwon KJ, Shin CY, Lee J, Han SH, Kim HY (2015) Depressive-like behaviors in a rat model of chronic cerebral hypoperfusion. Transl Stroke Res 6:207–214
Raghupathi R, Huh JW (2007) Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. J Neurotrauma 24:1596–1608
Ma J, Zhang K, Wang Z, Chen G (2016) Progress of research on diffuse axonal injury after traumatic brain injury. Neural Plast 2016:9746313. https://doi.org/10.1155/2016/9746313
Besenski N (2002) Traumatic injuries: imaging of head injuries. Eur Radiol 12:1237–1252
Gentry LR (1994) Imaging of closed head injury. Radiology 191:1–17
Hammoud DA, Wasserman BA (2002) Diffuse axonal injuries: pathophysiology and imaging. Neuroimaging Clin N Am 12:205–216
Zimmerman RA, Bilaniuk LT, Hackney DB, Goldberg HI, Grossman RI (1986) Head injury: early results of comparing CT and high-field MR. AJR Am J Roentgenol 147:1215–1222
Yanagawa Y, Tsushima Y, Tokumaru A, Un-no Y, Sakamoto T, Okada Y, Nawashiro H, Shima K (2000) A quantitative analysis of head injury using T2*-weighted gradient-echo imaging. J Trauma 49:272–277
Tong KA, Ashwal S, Holshouser BA, Shutter LA, Herigault G, Haacke AM, Kido DK (2003) Hemorrhagic shearing lesions in children and adolescents with posttraumatic diffuse axonal injury: improved detection and initial results. Radiology 227:332–339
Chan JH, Tsui EY, Peh WC, Fong D, Fok KF, Leung KM, Yuen MK, Fung KK (2003) Diffuse axonal injury: detection of changes in anisotropy of water diffusion by diffusion-weighted imaging. Neuroradiology 45:34–38
Takayama H, Kobayashi M, Sugishita M, Mihara B (2000) Diffusion-weighted imaging demonstrates transient cytotoxic edema involving the corpus callosum in a patient with diffuse brain injury. Clin Neurol Neurosurg 102:135–139
Ezaki Y, Tsutsumi K, Morikawa M, Nagata I (2006) Role of diffusion-weighted magnetic resonance imaging in diffuse axonal injury. Acta Radiol 47:733–740
Huisman TA, Sorensen AG, Hergan K, Gonzalez RG, Schaefer PW (2003) Diffusion-weighted imaging for the evaluation of diffuse axonal injury in closed head injury. J Comput Assist Tomogr 27:5–11
Arfanakis K, Haughton VM, Carew JD, Rogers BP, Dempsey RJ, Meyerand ME (2002) Diffusion tensor MR imaging in diffuse axonal injury. AJNR Am J Neuroradiol 23:794–802
Huisman TA, Schwamm LH, Schaefer PW, Koroshetz WJ, Shetty-Alva N, Ozsunar Y, Wu O, Sorensen AG (2004) Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol 25:370–376
Budde MS, Kim JH, Liang HF, Schmidt RE, Russell JH, Cross AH, Song SK (2007) Toward accurate diagnosis of white matter pathology using diffusion tensor imaging. Magn Reson Med 57:688–695
Donald CLM, Dikranian K, Song SK, Bayly PV, Holtzman DM, Brody DL (2007) Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol 2005:116–131
An C, Jiang X, Pu H, Hong D, Zhang W, Hu X, Gao Y (2016) Severity-dependent long-term spatial learning-memory impairment in a mouse model of traumatic brain injury. Transl Stroke Res 7:512–520
Joseph MJ, Caliaperumal J, Schlichter LC (2016) After intracerebral hemorrhage, oligodendrocyte precursors proliferate and differentiate inside white-matter tracts in the rat striatum. Transl Stroke Res 7:192–208
Tomita K, Nakada TA, Oshima T, Motoshima T, Kawaguchi R, Oda S (2019) Tau protein as a diagnostic marker for diffuse axonal injury. PLoS ONE 14. https://doi.org/10.1371/journal.pone.0214381
Lioutas VA, Alfaro-Martinez F, Bedoya F, Chung CC, Pimentel DA, Novak V (2015) Intranasal insulin and insulin-like growth factor 1 as neuroprotectants in acute ischemic stroke. Transl Stroke Res 6:264–275
Mammis A, McIntosh TK, Maniker AH (2009) Erythropoietin as a neuroprotective agent in traumatic brain injury Review. Surg Neurol 71:527–531
Kato K, Ishiguro Y, Suzuki F, Ito A, Semba R (1982) Distribution of nervous system-specific forms of enolase in peripheral tissues. Brain Res 237:441–448
Lamers KJ, van Engelen BG, Gabreëls FJ, Hommes OR, Borm GF, Wevers RA (1995) Cerebrospinal neuron-specific enolase, S-100 and myelin basic protein in neurological disorders. Acta Neurol Scand 92:247–251
Persson L, Hårdemark HG, Gustafsson J, Rundström G, Mendel-Hartvig I, Esscher T, Påhlman S (1987) S-100 protein and neuron-specific enolase in cerebrospinal fluid and serum: markers of cell damage in human central nervous system. Stroke 18:911–918
Raabe A, Grolms C, Keller M, Döhnert J, Sorge O, Seifert V (1998) Correlation of computed tomography findings and serum brain damage markers following severe head injury. Acta Neurochir 140:787–792
Raabe A, Menon DK, Gupta S, Czosnyka M, Pickard JD (1998) Jugular venous and arterial concentrations of serum S-100B protein in patients with severe head injury: a pilot study. J Neurol Neurosurg Psychiatry 65:930–932
Rothoerl RD, Woertgen C, Holzschuh M, Metz C, Brawanski A (1998) S-100 serum levels after minor and major head injury. J Trauma 45:765–767
Skogseid IM, Nordby HK, Urdal P, Paus E, Lilleaas F (1992) Increased serum creatine kinase BB and neuron specific enolase following head injury indicates brain damage. Acta Neurochir 115:106–111
Usui A, Kato K, Abe T, Murase M, Tanaka M, Takeuchi E (1989) S-100ao protein in blood and urine during open-heart surgery. Clin Chem 35:1942–1944
Woertgen C, Rothoerl RD, Holzschuh M, Metz C, Brawanski A (1997) Comparison of serial S-100 and NSE serum measurements after severe head injury. Acta Neurochir 139:1161–1164
Yamakami I, Yamaura A, Isobe K (1993) Types of traumatic brain injury and regional cerebral blood flow assessed by 99mTc-HMPAO SPECT. Neurol Med Chir (Tokyo) 33:7–12
Yamazaki Y, Yada K, Morii S, Kitahara T, Ohwada T (1995) Diagnostic significance of serum neuron-specific enolase and myelin basic protein assay in patients with acute head injury. Surg Neurol 43:267–270
Pleines UE, Morganti-Kossmann MC, Rancan M, Joller H, Trentz O, Kossmann T (2001) S-100 beta reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J Neurotrauma 18:491–498
de Kruijk JR, Leffers P, Menheere PP, Meerhoff S, Twijnstra A (2001) S-100B and neuron-specific enolase in serum of mild traumatic brain injury patients. A comparison with health controls. Acta Neurol Scand 103:175–179
Savola O, Pyhtinen J, Leino TK, Siitonen S, Niemelä O, Hillbom M (2004) Effects of head and extracranial injuries on serum protein S100B levels in trauma patients. J Trauma 56:1229–1234
Olsson A, Csajbok L, Ost M, Höglund K, Nylén K, Rosengren L, Nellgård B, Blennow K (2004) Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol 251:870–876
Zhong YH, Zheng BE, He RH, Zhou Z, Zhang SQ, Wei Y, Fan JZ (2020) Serum levels of HDL cholesterol are associated with diffuse axonal injury in patients with traumatic brain injury. Neurocrit Care. https://doi.org/10.1007/s12028-020-01043-w
Liang Y, Tong F, Zhang L, Zhu L, Li W, Huang W, Zhao S, He G, Zhou Y (2019) iTRAQ-based proteomic analysis discovers potential biomarkers of diffuse axonal injury in rats. Brain Res Bull 153:289–304. https://doi.org/10.1016/j.brainresbull.2019.09.004
Zhang P, Zhu S, Zhao M, Zhao P, Zhao H, Deng J, Li J (2018) Identification of plasma biomarkers for diffuse axonal injury in rats by iTRAQ-coupled LC–MS/MS and bioinformatics analysis. Brain Res Bull 142:224–232. https://doi.org/10.1016/j.brainresbull.2018.07.015
Carney N, Totten AM, O'Reilly S, Ullman JS, Hawryluk GWJ, Bell MJ, Bratton SL, Chesnut R, Harris OA, Kissoon N, Rubiano AM, Shutter L, Tasker RC, Vavilala MS, Wilberger J, Wright DW, Ghajar J (2017) Guidelines for the management of severe traumatic brain injury, Fourth Edition. Neurosurgery 18:6–15
Okonkwo DO, Büki A, Siman R, Povlishock JT (1999) Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport 10:353–358
Okonkwo DO, Povlishock JT (1999) An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab 19:443–451
Staal JA, Dickson TC, Chung RS, Vickers JC (2007) Cyclosporin-A treatment attenuates delayed cytoskeletal alterations and secondary axotomy following mild axonal stretch injury. Dev Neurobiol 67:1831–1842
Buki A, Farkas O, Doczi T, Povlishock JT (2003) Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma 20:261–268
Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW, Wang KKW, Hall ED (2001) The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma 18:1229–1240
Lubisch W, Beckenbach E, Bopp S, Hofmann H-P, Kartal A, Kästel C, Lindner T, Metz-Garrecht M, Reeb J, Regner F, Vierling M, Möller A (2003) Benzoylalanine-derived ketoamides carrying vinylbenzyl amino residues: discovery of potent water-soluble calpain inhibitors with oral bioavailability. J Med Chem 46:2404–2412
Ma M, Li L, Wang X, Bull DL, Shofer FS, Meaney DF, Neumar RW (2012) Short-duration treatment with the calpain inhibitor mdl-28170 does not protect axonal transport in an in vivo model of traumatic axonal injury. J Neurotrauma 29:445–451. https://doi.org/10.1089/neu.2011.2060
Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR, McIntosh TK (1996) Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A 93:3428–3433
Marmarou CR, Povlishock JT (2006) Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury. Exp Neurol 197:353–362
Reeves TM, Phillips LL, Lee NN, Povlishock JT (2007) Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain Res 1154:225–236
Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT (2001) The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma 18:607–614
Suehiro E, Singleton RH, Stone JR, Povlishock JT (2001) The immunophilin ligand FK506 attenuates the axonal damage associated with rapid rewarming following posttraumatic hypothermia. Exp Neurol 172:199–210
Omelchenko A, Shrirao AB, Bhattiprolu AK, Zahn JD, Schloss RS, Dickson S, Meaney DF, Boustany NN, Yarmush ML, Firestein BL (2019) Dynamin and reverse-mode sodium calcium exchanger blockade confers neuroprotection from diffuse axonal injury. Cell Death Dis 10. https://doi.org/10.1038/s41419-019-1908-3
Cross DJ, Garwin GG, Cline MM, Richards TL, Yarnykh V, Mourad PD, Ho RJ, Minoshima S (2015) Paclitaxel improves outcome from traumatic brain injury. Brain Res 1618:299–308. https://doi.org/10.1016/j.brainres.2015.06.006
Kleele T, Marinković P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F, Bishop D, Herms J, Konnerth A, Kerschensteiner M, Godinho L, Misgeld T (2014) An assay to image neuronal microtubule dynamics in mice. Nat Commun 5:4827. https://doi.org/10.1038/ncomms5827
Baas PW, Ahmad FJ (2013) Beyond taxol: microtubule-based treatment of disease and injury of the nervous system. Brain 136:2937–2951
Brizuela M, Blizzard CA, Chuckowree JA, Dawkins E, Gasperini RJ, Young KM, Dickson TC (2015) The microtubule-stabilizing drug Epothilone D increases axonal sprouting following transection injury in vitro. Mol Cell Neurosci 66:129–140
Ruschel J, Bradke F (2017) Systemic administration of epothilone D improves functional recovery of walking after rat spinal cord contusion injury. Exp Neurol. https://doi.org/10.1016/j.expneurol.2017.12.001
Fujita M, Oda Y, Wei EP, Povlishock JT (2011) The combination of either tempol or FK506 with delayed hypothermia: implications for traumatically induced microvascular and axonal protection. J Neurotrauma 28:1209–1218
Oda Y, Gao G, Wei EP, Povlishock JT (2011) Combinational therapy using hypothermia and the immunophilin ligand FK506 to target altered pial arteriolar reactivity, axonal damage, and blood-brain barrier dysfunction after traumatic brain injury in rat. J Cereb Blood Flow Metab 31:1143–1154
Clifton GL, Coffey CS, Fourwinds S, Zygun D, Valadka A, Smith KR Jr, Frisby ML, Bucholz RD, Wilde EA, Levin HS, Okonkwo DO (2012) Early induction of hypothermia for evacuated intracranial hematomas: a post hoc analysis of two clinical trials. J Neurosurg 117:714–720
Margulies S, Hicks R (2009) Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma 26:925–939
Smith DH, Hicks R, Povlishock JT (2013) Therapy development for diffuse axonal injury. J Neurotrauma 30:307–322
Yatsiv I, Grigoriadis N, Simeonidou C, Stahel PF, Schmidt OI, Alexandrovitch AG, Tsenter J, Shohami E (2005) Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J 19:1701–1703
Assandri R, Egger M, Gassmann M, Niggli E, Bauer C, Forster I, Gorlach A (1999) Erythropoietin modulates intracellular calcium in a human neuroblastoma cell line. J Physiol 516:343–352
Tubbs RS, Shoja MM, Jamshidi M, Shokouhi G (2007) Does the neuroprotective agent erythropoietin amplify diffuse axonal injury in its early stages? Med Hypotheses 69:1385
Benoit JS, Hannay HJ, Yamal J-M, Francis DH, Aisiku I, Robertson C (2019) Longitudinal Changes in disability rating scale scores: a secondary analysis among patients with severe TBI enrolled in the Epo Clinical Trial. J Int Neuropsychol Soc 25:293–301
Robertson CS, Hannay HJ, Yamal J-M, Gopinath S, Goodman JC, Tilley BC, Investigators ESTT (2014) Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA 312:36–47
Djebaili M, Hoffman SW, Stein DG (2004) Allopregnanolone and progesterone decrease cell death and cognitive deficits after a contusion of the rat pre-frontal cortex. Neuroscience 123:349–359
O'Connor CA, Cernak I, Johnson F, Vink R (2007) Effects of progesterone on neurologic and morphologic outcome following diffuse traumatic brain injury in rats. Exp Neurol 205:145–153
Xiao G, Wei J, Yan W, Wang W, Lu Z (2008) Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care 12:R61
Lu XY, Sun H, Li QY, Lu PS (2016) Progesterone for traumatic brain injury: a meta-analysis review of randomized controlled trials. World Neurosurg 90:199–210
Li HJ, Sun ZL, Pan YB, Xu MH, Feng DF (2019) Effect of α7nAChR on learning and memory dysfunction in a rat model of diffuse axonal injury. Exp Cell Res 383. https://doi.org/10.1016/j.yexcr.2019.111546
Liska MG, Crowley MG, Nguyen H, Borlongan CV (2017) Biobridge concept in stem cell therapy for ischemic stroke. J Neurosurg Sci 61:173–179
Riess P, Molcanyi M, Bentz K, Maegele M, Simanski C, Carlitscheck C, Schneider A, Hescheler J, Bouillon B, Schäfer U, Neugebauer E (2007) Embryonic stem cell transplantation after experimental traumatic brain injury dramatically improves neurological outcome, but may cause tumors. J Neurotrauma 24:216–225
Boockvar JA, Schouten J, Royo J, Millard M, Spangler Z, Castelbuono D, Snyder E, O’Rourke D, McIntosh T (2005) Experimental traumatic brain injury modulates the survival, migration, and terminal phenotype of transplanted epidermal growth factor receptor-activated neural stem cells. Neurosurgery 56:163–171
Erdö F, Bührle C, Blunk J, Hoehn M, Xia Y, Fleischmann B, Föcking M, Küstermann E, Kolossov E, Hescheler J, Hossmann KA, Trapp T (2003) Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. J Cereb Blood Flow Metab 23:780–785
Gao J, Prough DS, McAdoo DJ, Grady JJ, Parsley MO, Ma L, Tarensenko YI, Wu P (2006) Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol 201:281–292
Mahmood A, Lu D, Yi L, Chen JL, Chopp M (2001) Intracranial bone marrow transplantation after traumatic brain injury improving functional outcome in adult rats. J Neurosurg 94:589–595
Shear DA, Tate MC, Archer DR, Hoffman SW, Hulce VD, Laplaca MD, Stein DG (2004) Neural progenitor cell transplants promote long-term functional recovery after traumatic brain injury. Brain Res 1026:11–22
Maruichi K, Kuroda S, Chiba Y, Hokari M, Shichinohe H, Hida K, Iwasaki Y (2009) Transplanted bone marrow stromal cells improves cognitive dysfunction due to diffuse axonal injury in rats. Neuropathology 29(4):422–32. https://doi.org/10.1111/j.1440-1789.2008.00995.x
Lu D, Li Y, Wang L, Chen J, Mahmood A, Chopp M (2001) Intraarterial administration of marrow stromal cells in a rat model of traumatic brain injury. J Neurotrauma 18:813–819
Mahmood A, Lu D, Qu C, Goussev A, Chopp M (2006) Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J Neurosurg 104:272–277
Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L (2002) Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 22:6570–6577
GrandPré T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417:547–551
Curry DJ, Wright DA, Lee RC, Kang JJ, Frim DM (2004) Surfactant poloxamer 188-related decreases in inflammation and tissue damage after experimental brain injury in rats. J Neurosurg 101:91–96
Mbye LH, Keles E, Tao L, Zhang J, Chung J, Larvie M, Koppula R, Lo EH, Whalen MJ (2012) Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 32:515–524
Serbest G, Horwitz J, Jost M, Barbee K (2006) Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J 20:308–310
Agoston DV, Vink R, Helmy A, Risling M, Nelson D, Prins M (2019) How to translate time: the temporal aspects of rodent and human pathobiological processes in traumatic brain injury. J Neurotrauma 36:1724–1737. https://doi.org/10.1089/neu.2018.6261
Volovici V, Steyerberg EW, Cnossen MC, Haitsma IK, Dirven CMF, Maas AIR, Lingsma HF (2019) Evolution of evidence and guideline recommendations for the medical management of severe traumatic brain injury. J Neurotrauma 36:3183–3189
Acknowledgments
We thank the scientific staff of the Medical Library of the Erasmus MC for aiding us in conducting the search and M.W.T. van Bilsen, MD, for the design and illustration of Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Comments
Traumatic axonal injury (TAI) is a broad and heterogeneous topic and still a difficult and hardly to treat condition. In this comprehensive and timely review, the authors summarize very well the underlying pathophysiology of TAI and its diagnostic and therapeutic options. After the initial trauma directly causing primary axotomy, prevention of secondary damage is paramount. Unfortunately, currently treatment options are sparse and primarily concern the management of local and systemic effects of the event, while no specific and tailored therapies for TAI are yet available. Therefore, often only clinical stabilization and damage control, and rehabilitative repair work and clean-up efforts after the devastating impact are left. Fortunately, a multitude of promising research aiming to identify potential therapies is being carried out worldwide. As the authors have already mentioned, two main treatment types can be identified. The first type includes therapies that focus on the prevention of secondary axotomy whereas the second type aims to enhance neuronal regeneration after injuries that have already occurred. While we continuously expand our knowledge about TAI from past, present and future studies, additionally more and more data will be forthcoming in support of management of patients with TAI. In this context, the present article is a valuable contribution to an important and continually evolving field and provides extensive insight for those involved in the diagnostics and treatment of patients suffering from brain trauma and TAI.
Markus F. Oertel
Zurich, Switzerland
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Brain Trauma
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bruggeman, G.F., Haitsma, I.K., Dirven, C.M.F. et al. Traumatic axonal injury (TAI): definitions, pathophysiology and imaging—a narrative review. Acta Neurochir 163, 31–44 (2021). https://doi.org/10.1007/s00701-020-04594-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-020-04594-1