
Abstract
In this paper, we report on the utilization of micro-technology based tools to fight viral infections. Inspired by various hemoperfusion and immune-affinity capture systems, a blood virus depletion device has been developed that offers highly efficient capture and removal of the targeted virus from the circulation, thus decreasing virus load. Single-domain antibodies against the Wuhan (VHH-72) virus strain produced by recombinant DNA technology were immobilized on the surface of glass micro-beads, which were then utilized as stationary phase. For feasibility testing, the virus suspension was flown through the prototype immune-affinity device that captured the viruses and the filtered media left the column. The feasibility test of the proposed technology was performed in a Biosafety Level 4 classified laboratory using the Wuhan SARS-CoV-2 strain. The laboratory scale device actually captured 120,000 virus particles from the culture media circulation proving the feasibility of the suggested technology. This performance has an estimated capture ability of 15 million virus particles by using the therapeutic size column design, representing three times over-engineering with the assumption of 5 million genomic virus copies in an average viremic patient. Our results suggested that this new therapeutic virus capture device could significantly lower virus load thus preventing the development of more severe COVID-19 cases and consequently reducing mortality rate.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the first hospitalizations in December 2019 [1], and the detection of the new coronavirus strain in January 2020 [2], the COVID-19 outbreak causes public healthcare and social crisis worldwide [3], and apparently remains for years [4]. During the first year of this pandemic, almost 100 million people got infected [5]. PCR methods and lateral flow tests are the most widely used diagnostic tools [6, 7], but CRISPR/CAS systems [8], antibody functionalized bioactive nanomaterials, and other technologies were also implemented to overcome this unseen global challenge [9,10,11]. Parallel to the diagnostic endeavors, new therapy strategies have been utilized and/or developed, such as antivirals, convalescent plasma treatments, antibody therapies, extracorporeal treatments, and virus depletion methods for clinical use. Furthermore, vaccine development against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was immensely accelerated [12] and as of the beginning of 2022, vaccination seems to be an effective way of immunization [13,14,15,16,17,18]. However, naturally occurring genetic viral mutations resulted in different variants prolonging the challenge [18].
Approximately 17–40% of SARS-CoV-2 positive patients reported no typical symptoms [19, 20] while on the other hand, about 15–30% of patients develop respiratory failure requiring hospital admission [21], 12% need ventilation, and 3% extracorporeal life support [14, 22]. Certain comorbidities such as hypertension, diabetes, and obesity are reportedly represent serious risk factors with poor prognosis [23,24,25]. Cardiovascular complications including thrombosis [26] and systemic, cytokine-mediated inflammation are also associated with severe SARS-CoV-2 infection. Most importantly, high viral load during viremia predicts poor survival [27,28,29]. Later in the disease course, approximately 2 weeks after the onset of the symptoms and initial viral replication in the upper respiratory tract, spreading progresses towards the lower respiratory tract and creates secondary viremia followed by an attack against ACE2-expressing tissues [30]. Spreading correlates with clinical deterioration, which usually occurs roughly 2 weeks after the onset of the symptoms. In addition, other organs possibly reached directly by the virus through the circulation during persisting or temporary viremia may also occur in severe COVID-19 cases [31,32,33]. High plasma viral loads indicate increased severity, potential organ damage, and high chance of mortality [27, 30, 34,35,36].
In general, separation and purification technologies are frequently studied and utilized for biomedical and clinical applications [37,38,39,40,41,42,43,44,45]. Extracorporeal membrane oxygenation and extracorporeal CO2 removal can be used to address respiratory failures. Furthermore, cytokine removal by continuous renal replacement therapy or direct hemoperfusion with specific filters, HA resin hemoperfusion cartridges, and Toraymixin polymixin-B endotoxin removal are available therapeutic strategies during cytokine storm caused by SARS-CoV-2 infection [13, 46, 47]. Early cytokine removal may prevent the progression of respiratory failure or other organ dysfunctions in critically ill patients [48, 49], extracorporeal treatments, applied in clinical practice in the case of COVID-19 intensive care units, are based on non-specific binding or the adsorption of molecules to a large specific surface area having sponge-like symmetric pore structure [47, 50,51,52].
Next to the hemofilters, hemoperfusion and plasmapheresis procedures, such as heparin-immobilized polyethylene beads, are also used for non-specific pathogen removal from the bloodstream. The attached heparin binds viruses, bacteria, fungi, and toxins similarly to heparane-sulfate interacting with cell surface [53, 54]. Hemopurifier lectin affinity plasmapheresis filters have also been designed for whole virus, exosome, and exosomal microRNA removal and investigated for COVID-19 therapy [55].
In this paper, we report a feasibility study for the development and implementation of a SARS-CoV-2 capture device to specifically remove virions from the circulation, preventing the development of serious clinical outcomes. The approach combines the specificity of affinity chromatography with the high throughput hemoperfusion technique to fight the coronavirus pandemic. A laboratory scale hemoperfusion system was designed and fabricated to study the feasibility of the proposed approach. The setup has been tested in a BSL-4 laboratory environment with live virus suspensions as representative conditions and models to explore any potential risk factors before further development.
Materials and methods
Chemicals
Picoline borane, DMEM cell culture media, and phosphate buffered saline (PBS, pH 7.4) were from Sigma-Aldrich (St Louis, MO). Polyethylene-glycol was purchased from Merck (Kenilworth, NJ). Fetal bovine serum, trypsin-EDTA, and 3-aminopropyltriethoxysilane (APTES) were from Thermo Fisher Scientific (Waltham, MA). Glutaraldehyde was purchased from Carl Roth Chemicals (Karlsruhe, Germany). Methanol and absolute ethanol were purchased from VWR (Radnor, PA). Hydrochloric acid, sulfuric acid, and dry toluene were purchased from Molar Chemicals Kft. (Halasztelek, Hungary). Kanamycin was from SERVA Electrophoresis GmbH (Heidelberg, Germany). Isopropyl β-D-1-thiogalactopyranoside (IPTG) was from Biosynth AG (Staad, Switzerland). The SDS PAGE gel was made from 40% acrylamide solution from Bio-Rad (Hercules, CA) and HPLC grade water in a ratio of 37.5:1. Buffer “A” contained NaH2PO4 from Merck (Kenilworth, NJ) and NaCl from VWR (Radnor, PA). Imidazole for buffer “B” was purchased from Merck.
Expression and purification of SARS-CoV-2 spike protein–specific single-domain antibody
The coding sequence of the publicly available single-domain antibody (sdAb) against SARS-CoV-2 spike protein was codon optimized for E. coli [56] and the protein expressed from plasmid pET28b in SHuffle T7 E. coli strain, followed by purification using Ni-affinity chromatography as published earlier [57, 58]. The details of the procedure can be found in the Electronic Supporting Material.
Immobilization of anti-SARS-CoV-2 sdAb onto the surface of glass beads
It should be noted that increased stress level of >150 Pa may cause extensive cell damage. To avoid any damage of the different blood constituents, a hydro-dynamically similar model system to an approved extracorporeal technology, i.e., Cytosorb (Berlin, Germany), was utilized. For this purpose, commercially available monodisperse 450 μm diameter soda-lime glass beads were obtained from Glass Sphere (Jablonec nad Nisou, Czech Republic) due to their available immobilization chemistry and biocompatibility (composition: SiO2 61–67%, Na2O 10–18%, CaO 5–10%, B2O3 1–5%, Al2O3 3–8%). The SARS-CoV-2 spike protein–specific sdAb was immobilized onto the surface of the beads as follows. First, the beads were treated with a 1:1 mixture of cc. hydrochloric acid and methanol for 30 min at room temperature, followed by washing with HPLC grade water. After the washing step, the beads were treated with cc. sulfuric acid for 30 min at room temperature. The beads were washed and treated by boiling HPLC grade water for 30 min [59] and dried in an oven at 100 °C for 40 min. The dried beads were shaken in 3% APTES in dry toluene for 2 h at room temperature. Then, the excess reagents were rinsed off by dry toluene and the beads were treated at 100 °C until complete drying. The beads were then shaken in 2% glutaraldehyde in HPLC water for 1 h at room temperature and the excess glutaraldehyde was removed by rinsing with HPLC grade water. 1 mg/ml picoline borane solution was prepared in 5% ethanol in HPLC water as coupling buffer, and then 100 μl of 80 μg/ml sdAb solution was added. The beads were shaken overnight in the coupling buffer at 4 °C followed by rinsing with PBS, and stored at 4 °C until further processing. Non-specific binding sites were blocked by incubating the beads in 10 mg/ml polyethylene-glycol (MW 8000) containing PBS at 4°C for four hours before use.
Virus propagation
SARS-CoV-2 (hCoV-19 / Hungary / SRC_isolate_2 / 2020, Accession ID: EPI_ISL_483637) virus isolate was used for the experiments. Propagation of the viruses was carried out in VeroE6 cells (African green monkey kidney epithelial, ATCC CRL-1586) cultured in DMEM cell culture media containing 10% heat-inactivated fetal bovine serum. Cells were incubated at 37 °C in humidified air supplied with 5% CO2.
Virus capture
All virus culturing and capture experiments were carried out in the facility of the National Virology Laboratory, University of Pecs, Hungary, which is a Biosafety Level 4 (BSL-4) facility fulfilling all the requirements of the US Centers for Disease Control and Prevention guide for Biosafety in Microbiological and Biomedical Laboratories (6th Edition) [60]. Shortly, the laboratory is located in a separate building having dedicated supply and exhaust ventilation equipment with vacuum and decontamination systems. Operators were wearing a full-body, air-supplied positive pressure suit, while −120 Pa pressure was maintained in the rooms. The utilized conditions aimed to prevent any aerosol mediated infections.
Standard stainless steel HPLC columns (length: 16.5 cm, I.D. 4.6 mm) were modified to obtain laboratory-scale test cartridges. Four hundred micrometer hole size meshes were placed in the Swagelok fittings to retain the beads during the circulation. Four grams of anti-SARS-CoV-2 sdAb immobilized beads was loaded into the cartridges for the virus capture experiment. The viruses, at the average of ~350 viral genome copies/μl concentration, were spiked into 30 ml DMEM medium and the virus suspension was circulated through the cartridges under investigation using a Masterflex peristaltic pump (Cole-Parmer, Vernon Hills, IL) for 2 h (i.e., 20 cycles in total). An open-lid vessel was inserted in the circulation system to equilibrate the fluid level and ensure bubble-free flow. Furthermore, the vessel served as injection and sampling point as well. The schematics and photo of the experimental setup are shown in Fig. 1. The virus suspension was continuously homogenized during the experiments using a magnetic stirrer. The flow rate was set to 5 ml/min (corresponding to 155 cm/min linear velocity). To monitor any non-specific binding of the virus particles, a control experiment was also performed. The setup of the control experiment was identical as described above, but only the linker (i.e., glutaraldehyde) was immobilized onto the bead surface, and the non-specific binding sites were masked by 10 mg/ml polyethylene-glycol (MW 8000) containing PBS as suggested by Monard et al. [50]. The virus suspension was circulated continuously for 2 h and after the circulation was stopped, the column was rinsed with PBS and the beads were transferred to falcon tubes. Virus particles were detached from the surface of the beads by washing with 1 ml 1x trypsin-EDTA solution in PBS for 5 min. Then, 2 ml PBS was added before further incubation for 5 min at 37°C. The bead-virus particle suspension was vortexed thoroughly, then the beads were allowed to sink and the supernatant was sampled (referred to as eluent).

The laboratory scale system with the test cartridge (panel A) and its schematic representation (panel B). The circulation was supported by a peristaltic pump (1). The parts of the system—cartridge (2), vessel (3)—were connected with polyethylene tubing. The virus suspension in the vessel was homogenized by a magnetic stirrer (4). The vessel served as sampling point for the initial and residual samples
Quantitative virus genome copy determination
Duplicate samples of 100 μl were taken from the vessel (referred to as initial and residual) as well as from the supernatants (referred to as eluent) to determine the initial virus concentration and the number of captured virion particles, respectively. The number of captured virus particles was quantitatively determined by ddPCR (QX200 Droplet Digital PCR, Bio-Rad) as published earlier [61, 62]. Shortly, ddPCR is based on water-oil emulsion droplet technology. A sample is fractionated into a certain number of droplets, and PCR amplification of the template molecules occurs in each individual droplet [63]. After PCR amplification, concentrations are determined using the Poisson distribution of fluorescent and non-fluorescent droplets [64]. The Bio-Rad QuantaSoft software was used to evaluate the results. Nucleic acid isolation from the samples was performed using a Monarch Total RNA Miniprep Kit (New England Biolabs, Ipswich, MA) according to the vendor’s instruction to quantify the viral RNA. Furthermore, conventional qPCR method was utilized for the detection of SARS-CoV-2 virions in the eluent [65].
Scanning electron microscopy
Captured virus particles were visualized using scanning electron microscopy (FEI/ThermoFisher Apreo S LoVac). Observation by STEM was carried out in transmission mode under high-vacuum with 30 kV accelerating voltage. Samples were observed without fixation by following a standard air-drying procedure [66]. UV-C inactivated virus stock was purchased from RoLink Biotechnology (Pecs, Hungary). First, the virus suspension was dried onto a carbon-stabilized formvar-coated side-grid (SFR, Toronto, Canada) at room temperature for 20 min to determine its morphology. Then, glass beads with captured virus particles on their surface were taken out from the stainless steel cartridge after a regular capture cycle. To remove any remaining residues from the culture media, beads were rinsed with HPLC grade water. Low-vacuum mode was used with 10 kV accelerating voltage to detect the virus on the surface of the glass beads.
Results and discussion
In this paper, we report a feasibility study, first approach towards implementing a novel method to fight SARS-CoV-2 pandemic. A high throughput extracorporeal device has been designed to specifically remove virions from the circulation with the goal to avoid serious clinical outcome. A laboratory scale hemoperfusion system has been fabricated to study the feasibility of the proposed approach, which combined the specificity of affinity chromatography with the high throughput of hemoperfusion. The scheme of the proposed virus capture technique is shown in Fig. 2.

Schematic representation of the virus capture mechanism. The immobilized nanobodies specifically bound the spike protein of SARS-CoV-2 particles thus selectively capture them from the blood stream. Peripheral blood flows through the microbead packed bed from left to right. The bed is retained by the mesh. All intact blood components can be returned to the circulation without any known and/or expected side effects
Capture efficiency was investigated by using an actual strain of SARS-CoV-2 in a BSL-4 classified laboratory. In the presented proof of concept experiments, the virus particles were captured from Dulbecco Modified Eagle Medium containing L-glutamine, sodium pyruvate, sodium bicarbonate, and phenol red indicator as described in the experimental section. Albeit, its complexity lags behind that of blood complexity, it can adequately model the virus capture efficiency from fluidic circulation. Virus concentrations were determined by ddPCR, both in the active and control experiments (Table 1).
As apparent from Table 1, some virus particles must have been non-specifically bound to the plastic components of the equipment (buffer vessel, tubing, and connectors) and on the surface of the glass beads, represented by the difference between the initial-residual+eluent. However, it should be emphasized that the surface-to-volume ratio of this laboratory scale experimental setup was significantly higher than that of in an actual hemoperfusion device; thus, non-specific binding was considered to be overrepresented. The measure of non-specific binding was determined by balancing the captured virus copies in the active and control experiments. Non-specific binding of virions or any proteinous macromolecules is governed by physical adsorption (or physisorption), in which van der Waals forces, hydrophobic interactions, electrostatic interactions, and hydrogen bonds play essential roles. Physisorption is resulted due to the interaction of electron configuration of the contacting atoms or molecules. In spite of the numerous technological advancements in the field of protein immobilization [67], non-specific binding remained a critical bottleneck due to the lack of fundamental understanding of the phenomenon. Consequently, non-specific binding could not be completely avoided only decreased; therefore, its relatively high apparent measure is not surprising [67, 68]. From the practical point of view, it is expected that non-specific binding will be less important in an actual clinical environment, as during the priming step (i.e., the process, which removes the storage solution and any air bubbles from the extracorporeal cartridge) human serum albumin will block most if not all non-specific binding sites.
Note, only those virus particles were involved in the evaluation, which remained in the circulation and did not bind non-specifically, i.e., not removed from the circulation. Considering the reported genomic virus concentrations (Table 1), the volume of the processed virus suspensions, the residual and the eluted samples (30 ml, 30 ml, and 3 ml, respectively), the following absolute virus copies were counted: 10,425,000 (initial); 778,500 (residual); and 369,000 (eluent) in the case of the active experiment and 10,500,000, 3,150,000, and 247,900 in the case of the control. Thus, as a first approximation, we can conclude that the active cartridge removed 32% of the virus particles from the circulation, while the control cartridge was able to adsorb only 7%, apparently representing the level of non-specific binding. In other words, as both the active and control experiments also captured virions non-specifically; thus, only their differences were considered later as immuno-affinity captured virus particles, not the absolute values. The 4–5-fold difference in the residual virion copy number between the active and control experiments is notable, therefore suggests the feasibility of the proposed technology.
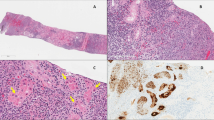
After the capture efficiency studies, scanning electron microscope (SEM) imaging was utilized to visualize the captured viruses on the surface of the beads. To this end, ~4·105 PFU/ml concentration virus suspension was dried onto a microscope slide-grid to observe certain morphologies of the virions under the conditions of the actual experimental setup (see Fig. 3A and B). Figure 3C and D show the captured virus particles on the surface of the beads. Please note that here the same capturing method was used as for the efficiency measurements above, but the captured virus particles were rinsed with HPLC grade water to remove any residual PBS in order to improve image sharpness. The resulted SEM images are apparently similar to recently published pictures made under similar conditions [69, 70]. After SEM imaging, the beads were trypsin-EDTA washed for verifying the microscopy observations. The existence of SARS-CoV-2 virions in the eluent was successfully verified by conventional qPCR method.

Scanning electron microscope (SEM) images of the virions. Panel (A): virus suspension dried onto a microscope slide-grid. Panel (B): single virion; the black patch visible in the center of the virus particle was caused by the electron beam. Panels (C) and (D): captured virus particles on the surface of the beads. Single and aggregated virions are indicated by black and white arrows, respectively. Aggregation of virus particles occurred due to the drying step of the sample preparation at room temperature
According to the current clinical protocols, similar extracorporeal treatments may last up to 24 h [71], that, based on our results, would readily capture practically all circulating virus particles. Both during the active and control experiments, the captured virus particles were eluted from the surface of the glass beads by buffer rinse with trypsin wash. Comparing the active and control experiments with the down-scaled laboratory model device filled with 2.6 cm3 of glass beads, the difference in the number of specifically captured viruses was considered to be the actual performance, i.e., found to be 120,000 virus copies. The captured 120,000 virus particles from the virus culture media circulation, suggested the partitioning ability of 15 million virus particles with an actual therapeutic size column. In other words, since the actual size of an average hemofiltration cartridge is about 330 cm3, the virus-binding capacity of our design is expected to be around 15 million. There is a reported correlation between human plasma SARS-CoV-2 virus concentration and COVID-19 disease course and mortality [27, 72]. Viremia is observed in 100%, 52.6%, and 11.1% of intensive care unit, non-intensive care unit, and outpatients of COVID-19 patients, respectively, and typical virus concentrations in the plasma of viremic patients range from 100 to 1000 copies/ml [73, 74]. Considering that an adult has an average of 5 l of blood, the amount of SARS-CoV-2 to be captured from the bloodstream at one time is expected to be around 5 million virus particles, so the system design reported in this paper holds the promise to have threefold excess capture capacity. Please note that the reported virus copy numbers in the literature represent the genomic concentrations, i.e., albeit the exact relationship between genomic virus concentration and infectious viral load level is still under debate [56, 75, 76]; all reported findings in this study should be interpreted from the technical feasibility point of view only. The actual therapeutic benefit of the proposed approach cannot be evaluated without comprehensive clinical trials. However, a very recent large cohort study indicated that the odds of mortality increased by 40% by each viremic day [77].
Please note the importance of future biocompatibility testing to shed light on any potential drawbacks of the proposed technology, which was not possible at this very early stage of the development. However, it is suspected that shear stress on blood elements, extracorporeal treatment–induced hypothermia, and blood exposure to non-biological surfaces may initiate perioperative systemic inflammatory response syndrome [78]. Another limitation of the method is the relatively fast appearance of various new mutated virus strains due to positive evolutionary selection, i.e., decreasing immune-affinity strength. In the presented particular case, the mutations resulted in the structure variation of the spike protein, which is targeted by the utilized sdAb for immune-affinity virus capture. Such mutations are very difficult to rapidly address from regulatory point of view, i.e., new antibody approvals, unless agencies authorize emergency use in case of urgent needs.
Conclusions
A laboratory scale virus capture device was designed and fabricated to evaluate the feasibility of a novel immuno-affinity capture–based extracorporeal treatment approach. Due to the recent SARS-CoV-2 pandemic, its Wuhan (VHH-72) virus strain was chosen as model system to probe the proposed technology. Single-domain antibodies were produced by recombinant DNA technology and immobilized on the surface of glass micro-beads. A laboratory size, i.e., down-scaled model device, was utilized for the feasibility tests. The experiments conducted in BSL4 laboratory environment using live virus suspensions successfully proved the virus capture ability of the reported method. In the lack of effective antiviral drugs, especially during the outbreak of a rear or unknown virus caused pandemic, the suggested technology may serve as the base of an effective extracorporeal therapy, which helps patients to fight against the infection and significantly decrease the chance of being admitted to intensive care units. Furthermore, it is anticipated that after further development, the reported technology could possibly be an artificial organ platform, which represent an option to address organ failure issues and improve survival in case of viremia caused by several types of viral infections.
References
Wu F et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269
Zhou P et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273
Haldane V et al (2021) Health systems resilience in managing the COVID-19 pandemic: lessons from 28 countries. Nat Med 27(6):964–980
Kissler SM, Tedijanto C (2020) Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 368(6493):860–868
Carvalho T, Krammer F, Iwasaki A (2021) The first 12 months of COVID-19: a timeline of immunological insights. Nat Rev Immunol 21(4):245–256
Weissleder R et al (2020) COVID-19 diagnostics in context. Sci Transl Med 12(546):eabc1931
Vandenberg O et al (2021) Considerations for diagnostic COVID-19 tests. Nat Rev Microbiol 19(3):171–183
Rahimi H et al (2021) CRISPR systems for COVID-19 diagnosis. ACS Sensors 6(4):1430–1445
Guo K et al (2021) Rapid single-molecule detection of COVID-19 and MERS antigens via nanobody-functionalized organic electrochemical transistors. Nat Biomed Eng 5(7):666–677
Tang Z et al (2020) A materials-science perspective on tackling COVID-19. Nat Rev Mater 5(11):847–860
Sheridan C (2020) COVID-19 spurs wave of innovative diagnostics. Nat Biotechnol 38(7):769–772
Mullard A (2020) COVID-19 vaccine development pipeline gears up. Lancet 395(10239):1751–1752
Swol J, Lorusso R (2020) Additive treatment considerations in COVID-19—the clinician’s perspective on extracorporeal adjunctive purification techniques. Artif Organs 44(9):918–925
Xi Y (2020) Convalescent plasma therapy for COVID-19: a tried-and-true old strategy? Signal Transduct Target Ther 5(1):203
Subbarao K, Mordant F, Rudraraju R (2020) Convalescent plasma treatment for COVID-19: tempering expectations with the influenza experience. Eur J Immunol 50(10):1447–1453
Jeyanathan M et al (2020) Immunological considerations for COVID-19 vaccine strategies. Nat Rev Immunol 20(10):615–632
Graham BS (2020) Rapid COVID-19 vaccine development. Science 368(6494):945–946
Tao K, Tzou PL, Nouhin J, Gupta RK, de Oliveira T, Kosakovsky Pond SL, Fera D, Shafer RW (2021) The biological and clinical significance of emerging SARS-CoV-2 variants. Nat Rev Genet 22(12):757–773. https://doi.org/10.1038/s41576-021-00408-x
Rasmussen AL, Popescu SV (2021) SARS-CoV-2 transmission without symptoms. Science 371(6535):1206–1207
Ma Q et al (2021) Global percentage of asymptomatic SARS-CoV-2 infections among the tested population and individuals with confirmed COVID-19 diagnosis: a systematic review and meta-analysis. JAMA Netw Open 4(12):e2137257
Ginestra JC et al (2022) COVID-19 critical illness: a data-driven review. Annu Rev Med 73:95–111
Pfortmueller CA et al (2021) COVID-19-associated acute respiratory distress syndrome (CARDS): current knowledge on pathophysiology and ICU treatment - a narrative review. Best Pract Res Clin Anaesthesiol 35(3):351–368
Chang MC et al (2020) Risk factors for disease progression in COVID-19 patients. BMC Infect Dis 20(1):445
Ayres JS (2020) A metabolic handbook for the COVID-19 pandemic. Nat Metab 2(7):572–585
Richardson S et al (2020) Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 323(20):2052–2059
Varga Z et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418
Fajnzylber J et al (2020) SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat Commun 11(1):5493
Pujadas E et al (2020) SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir Med 8(9):e70
Tsukagoshi H et al (2021) Relationships between viral load and the clinical course of COVID-19. Viruses 13(2):304
Cao W, Li T (2020) COVID-19: towards understanding of pathogenesis. Cell Res 30(5):367–369
Müller JA et al (2021) SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat Metab 3(2):149–165
Perotti C, Del Fante C (2020) Plasma from donors recovered from the new Coronavirus 2019 as therapy for critical patients with COVID-19 (COVID-19 plasma study): a multicentre study protocol. Intern Emerg Med 15(5):819–824
Xu Z et al (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8(4):420–422
Hagman K, Hedenstierna M, Gille-Johnson P, Hammas B, Grabbe M, Dillner J, Ursing J (2020) SARS-CoV 2 RNA in serum as predictor of severe outcome in COVID-19: a retrospective cohort study. Clin Infect Dis 73(9):e2995–e3001. https://doi.org/10.1093/cid/ciaa1285
Eberhardt KA, Meyer-Schwickerath C, Heger E, Knops E, Lehmann C, Rybniker J (2020) RNAemia corresponds to disease severity and antibody response in hospitalized COVID-19 patients. Viruses 12(9):1045. https://doi.org/10.3390/v12091045
Xu D et al (2021) Relationship between serum severe acute respiratory syndrome coronavirus 2 nucleic acid and organ damage in coronavirus 2019 patients: a cohort study. Clin Infect Dis 73(1):68–75
Stiapis CS, Skouras ED, Burganos VN (2020) Prediction of toxin removal efficiency of novel hemodialysis multilayered mixed-matrix membranes. Sep Purif Technol 250:117272
Westphalen H et al (2021) Assessment of hemodialysis clinical practices using polyaryl ether sulfone-polyvinylpyrrolidone (PAES: PVP) clinical membrane: modeling of in vitro fibrinogen adsorption, in situ synchrotron-based imaging, and clinical inflammatory biomarkers investigations. Sep Purif Technol 259:118136
Liu J et al (2020) Alginate/HSA double-sided functional PVDF multifunctional composite membrane for bilirubin removal. Sep Purif Technol 252:117295
Fu C-C et al (2020) Adsorptive removal of p-cresol and creatinine from simulated serum using porous polyethersulfone mixed-matrix membranes. Sep Purif Technol 245:116884
Malik DJ et al (2009) Synthesis and characterization of size-selective nanoporous polymeric adsorbents for blood purification. Sep Purif Technol 66(3):578–585
Basauri A et al (2021) Integrated strategy for the separation of endotoxins from biofluids. LPS capture on newly synthesized protein. Sep Purif Technol 255:117689
Dehghan R, Barzin J (2021) Membrane patterning through horizontally aligned microchannels developed by sulfated chopped carbon fiber for facile permeability of blood plasma components in low-density lipoprotein apheresis. Sep Purif Technol 278:119512
Haghdoost F et al (2021) Preparation and characterization of electrospun polyethersulfone/polyvinylpyrrolidone-zeolite core–shell composite nanofibers for creatinine adsorption. Sep Purif Technol 257:117881
Guzman NA, Guzman DE (2021) Immunoaffinity capillary electrophoresis in the era of proteoforms, liquid biopsy and preventive medicine: a potential impact in the diagnosis and monitoring of disease progression. Biomolecules 11(10):1443. https://doi.org/10.3390/biom11101443
Barbaro RP et al (2021) Extracorporeal membrane oxygenation for COVID-19: evolving outcomes from the international Extracorporeal Life Support Organization Registry. Lancet 398(10307):1230–1238
Ronco C et al (2021) Extracorporeal blood purification and organ support in the critically ill patient during COVID-19 pandemic: expert review and recommendation. Blood Purif 50(1):17–27
Ramírez-Guerrero G et al (2021) Early cytokine removal in critical COVID-19 patients with extracorporeal therapies (HA-380 plus high volume hemofiltration) may prevent progression of acute respiratory distress syndrome: case report. Blood Purif 50(4-5):575–577
Shadvar K et al (2021) Hemoperfusion as a potential treatment for critically ill COVID-19 patients with cytokine storm. Blood Purif 50(3-4):405–407
Monard C, Abraham P, Schneider A, Rimmelé T (2022) New targets for extracorporeal blood purification therapies in sepsis. Blood Purif 52(1):1–7. https://doi.org/10.1159/000524973
Monard C, Rimmelé T, Ronco C (2019) Extracorporeal blood purification therapies for sepsis. Blood Purif 47(3):1–14. https://doi.org/10.1159/000499520
Ankawi G et al (2018) Extracorporeal techniques for the treatment of critically ill patients with sepsis beyond conventional blood purification therapy: the promises and the pitfalls. Crit Care 22(1):262
Seffer MT et al (2021) Heparin 2.0: a new approach to the infection crisis. Blood Purif 50(1):28–34
Pape A et al (2021) Treatment of a critically ill COVID-19 patient with the Seraph 100 Microbind Affinity filter. TH Open 05(02):e134–e138
Amundson DE, Shah US, de Necochea-Campion R, Jacobs M, LaRosa SP, Fisher CJ (2021) Removal of COVID-19 spike protein, whole virus, exosomes, and exosomal MicroRNAs by the Hemopurifier® lectin-affinity cartridge in critically Ill patients with COVID-19 infection. Front Med 8:744141. https://doi.org/10.3389/fmed.2021.744141
Wrapp D et al (2020) Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 181(5):1004–1015.e15
Reider B et al (2021) Integrated workflow for urinary prostate specific antigen N-glycosylation analysis using sdAb partitioning and downstream capillary electrophoresis separation. Anal Chim Acta 1184:338892
Ren G, Ke N, Berkmen M (2016) Use of the SHuffle strains in production of proteins. Curr Protoc Protein Sci 85:5.26.1–5.26.21
Cras JJ et al (1999) Comparison of chemical cleaning methods of glass in preparation for silanization. Biosens Bioelectron 14(8):683–688
Meechan PJ, Potts J (2020) Biosafety in microbiological and biomedical laboratories 6th edition. U.S. Dept. of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institutes of Health, Washington D.C
Földes F, Madai M, Papp H, Kemenesi G, Zana B, Geiger L, Gombos K, Somogyi B, Bock-Marquette I, Jakab F (2020) Small interfering RNAs are highly effective inhibitors of Crimean-Congo hemorrhagic fever virus replication in vitro. Molecules 25(23):5771. https://doi.org/10.3390/molecules25235771
Barsi S et al (2022) Computational drug repurposing against SARS-CoV-2 reveals plasma membrane cholesterol depletion as key factor of antiviral drug activity. PLoS Comput Biol 18(4):e1010021
Hindson BJ et al (2011) High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83(22):8604–8610
Kojabad AA et al (2021) Droplet digital PCR of viral DNA/RNA, current progress, challenges, and future perspectives. J Med Virol 93(7):4182–4197
Bereczki I, Papp H, Kuczmog A, Madai M, Nagy V, Agócs A, Batta G, Milánkovits M, Ostorházi E, Mitrović A, Kos J, Zsigmond Á, Hajdú I, Lőrincz Z, Bajusz D, Keserű GM, Hodek J, Weber J, Jakab F et al (2021) Natural apocarotenoids and their synthetic glycopeptide conjugates inhibit SARS-CoV-2 replication. Pharmaceuticals (Basel) 14(11). https://doi.org/10.3390/ph14111111
Adams JR, Wilcox TA (1982) Scanning electron microscopical comparisons of insect virus occlusion bodies prepared by several techniques. J Invertebr Pathol 40:12–20
Frutiger A et al (2021) Nonspecific binding—fundamental concepts and consequences for biosensing applications. Chem Rev 121(13):8095–8160
Brambilla D, Mussida A, Ferretti AM, Sola L, Damin F, Chiari M (2022) Polymeric coating of silica microspheres for biological applications: suppression of non-specific binding and functionalization with biomolecules. Polymers (Basel) 14(4). https://doi.org/10.3390/polym14040730
Ehre C (2020) SARS-CoV-2 infection of airway cells. N Engl J Med 383(10):969
Caldas LA et al (2020) Ultrastructural analysis of SARS-CoV-2 interactions with the host cell via high resolution scanning electron microscopy. Sci Rep 10(1):16099
Peng JY, Li L, Zhao X, Ding F, Hou X, Peng Z (2021) Hemoperfusion with CytoSorb® in critically Ill COVID-19 patients. Blood Purif 51(5):410–416. https://doi.org/10.1159/000517721
Vivanti AJ et al (2020) Transplacental transmission of SARS-CoV-2 infection. Nat Commun 11(1):3572
Colagrossi L et al (2021) SARS-CoV-2 RNA in plasma samples of COVID-19 affected individuals: a cross-sectional proof-of-concept study. BMC Infect Dis 21(1):184
Jacobs JL et al (2022) Severe acute respiratory syndrome coronavirus 2 viremia is associated with coronavirus disease 2019 severity and predicts clinical outcomes. Clin Infect Dis 74(9):1525–1533
van Doremalen N, Purushotham JN, Schulz JE, Holbrook MG, Bushmaker T, Carmody A, Port JR, Yinda CK, Okumura A, Saturday G, Amanat F, Krammer F, Hanley PW, Smith BJ, Lovaglio J, Anzick SL, Barbian K, Martens C, Gilbert SC et al (2021) Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces viral shedding after SARS-CoV-2 D614G challenge in preclinical models. Sci Transl Med 13(607):eabh0755. https://doi.org/10.1126/scitranslmed.abh0755
Israelow B et al (2021) Adaptive immune determinants of viral clearance and protection in mouse models of SARS-CoV-2. Sci Immunol 6(64):eabl4509
Hagman K et al (2022) Duration of SARS-CoV-2 viremia and its correlation to mortality and inflammatory parameters in patients hospitalized for COVID-19: a cohort study. Diagn Microbiol Infect Dis 102(3):115595
Alarie M et al (2022) Use of CytoSorb® hemoadsorption column during prolonged cardiopulmonary bypass in complex cardiac surgery patient. J Cardiothorac Surg 17(1):172
Acknowledgements
This is contribution #197 of the Horváth Csaba Memorial Laboratory of Bioseparation Sciences. The authors acknowledge the kind help of Miklos Jakab of University of Pannonia with the SEM measurements.
Funding
Open access funding provided by University of Pannonia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
G.J. and A.G. are shareholders of CAPTEC Medical Ltd. CAPTEC did not have any specific role in the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript, should be disclosed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 30 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jarvas, G., Szerenyi, D., Jankovics, H. et al. Microbead-based extracorporeal immuno-affinity virus capture: a feasibility study to address the SARS-CoV-2 pandemic. Microchim Acta 190, 95 (2023). https://doi.org/10.1007/s00604-023-05671-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-023-05671-9