
Abstract
Currently, the determination of DNA methylation is still a challenge due to the limited efficiency of enrichment, bisulfite modification, and detection. In this study, a dual-modality loop-mediated isothermal amplification integrated with magnetic bead isolation is proposed for the determination of methylated Septin9 gene in colorectal cancer. Magnetic beads modified with anti-methyl cytosine antibody were prepared for fast enrichment of methylated DNA through specific immunoaffinity (30 min). One-pot real-time fluorescence and colorimetric loop-mediated isothermal amplification were simultaneously developed for detecting methylated Septin9 gene (60 min). The real-time fluorescence generating by SYTO-9 dye (excitation: 470 nm and emission: 525 nm) and pH indicator (neutral red) was used for quantitative and visualized detection of methylated DNA. This method was demonstrated to detect methylated DNA from HCT 116 cells ranging from 2 to 0.02 ng/μL with a limit of detection of 0.02 ± 0.002 ng/μL (RSD: 9.75%). This method also could discriminate methylated Septin9 in 0.1% HCT 116 cells (RSD: 6.60%), suggesting its high specificity and sensitivity. The feasibility of this assay was further evaluated by clinical plasma samples from 20 colorectal cancer patients and 20 healthy controls, which shows the potential application in simple, low cost, quantitative, and visualized detection of methylated nucleic acids.
Graphical abstract

A dual-modality loop-mediated isothermal amplification (LAMP) integrated with immuno-magnetic beads (IMB) enrichment was proposed for the determination of methylated Septin9 gene in colorectal cancer (CRC).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA methylation is a key epigenetic variation caused by a methyl group transferring to a cytosine residue at the 5′end of CpG dinucleotide (5′-CG-3′), named as 5-methylcytosine (5mC) [1,2,3,4]. Alterations in DNA methylation that trigger the onset of diseases such as cancer [5,6,7,8] are used for clinical diagnostics [9,10,11,12,13]. The current methods for detecting DNA methylation are typically including bisulfite conversion or restriction endonuclease-specific cleavage coupled with polymerase chain reaction (PCR) amplification or/and genomic sequencing [14, 15]. Bisulfite modification is a simple and mature approach but often suffers from the incomplete conversion or degradation of DNA due to the high GC content of target or the strong concentration of bisulfite. Enzymatic treatment is using restriction endonucleases such as HpaII to catalyze the digestion of unmethylated DNA effectively, while these enzymes are limited for some specific base sequences (e.g., CCGG) [16, 17]. Methylation-specific PCR needs a complex primer design according to the different methylation levels [18, 19]. Sequencing can accurately and directly identify single nucleotide base; however, it requires relatively high DNA concentration and purity as well as relies on professional experimental operation and expensive equipment [20]. Therefore, it is highly desirable to develop a simple, efficient, and universal method for the detection of low-abundance methylated DNA (mDNA).
Loop-mediated isothermal amplification (LAMP) is a powerful alternative to PCR, which enables simple, low cost, rapid, and sensitive detection of nucleic acids at a constant temperature (e.g., 60–65 °C), showing great application in clinical detection because of its good performance to crudely processed samples [21,22,23]. Real-time fluorescence LAMP is capable of real-time monitoring and sensitive quantitative detection by using fluorescent dyes such as SYTO-9 and SYBR Green. Colorimetric LAMP takes advantage of some colorimetric indicators such as metal ion indicators (e.g., calcein and Mn2+) and pH-sensitive dyes (e.g., neutral red) to sense the products of isothermal reaction in a direct visualization way without any readout equipment [24], showing great potential in the application of point-of-care diagnosis [24,25,26]. Colorimetric-based LAMP methods have been developed in a wide range of applications such as Zika virus detection [27], SNP genotyping [24], and SARS-CoV-2 detection [28, 29].
Magnetic beads (MB)-based immunorecognition has become an attractive approach for the rapid capture and isolation of specific analytes such as circulating tumor cell [30], exosome [31, 32], and DNA [33,34,35,36]. MB-based immuno-separation and enrichment offer a great number of advantages: simplicity, high efficiency, low cost, non-destruction, fast magnetic response capability, and good anti-interference ability. Therefore, compared with bisulfite conversion or restriction endonuclease-specific cleavage, MB-based strategy is more superior for the development of fast, easy, and effective method for isolation and enrichment of mDNA, which will help to improve sample recovery efficiency, release labor-intensive operations, and promote clinical application.
Based on the advantages of LAMP and MB which combines rapid, convenient, and low cost of nucleic acid enrichment and detection, herein, anti-methyl cytosine antibody (anti-5mC antibody) modified MB (immuno-MB, IMB) enrichment-assisted dual-modality LAMP was constructed for DNA methylation detection. IMB was fabricated to quickly isolate mDNA from DNA extracts through the specific binding between mDNA and anti-5mC antibody. Two signal reporting strategies, real-time fluorescent and colorimetric modality LAMP, were developed for simple, low cost, and user-friendly detection of specific mDNA in a single-step reaction. The proposed dual-modality LAMP signal readout enables the assay to be applied in different settings with high accuracy and flexibility.
Material and methods
Materials and reagents
Information about all the reagents, cultured cell, and nucleic acid sequences are included in the electronic supplementary material.
Instruments and characterization
Real-time fluorescence LAMP assay was performed on LineGene 9640 real-time nucleic acids amplification detection system (Hangzhou Bioer Technology Co., Ltd. Hangzhou, China). The concentration of nucleic acids was measured by SMA4000 UV–Vis Spectrophotometer (Meriton Ltd. Beijing, China). Electrophoresis instrument (Tanon EPS 300, Shanghai, China) was used in the analysis of nucleic acid amplification products. The gels were imaged by the imaging system (Tanon-4100). Zeta potential was measured by ZetaSizer Nano ZS90 (Malvern Instruments, Worcestershire, UK). To obtain the zeta potential of MB, IMB, and IMB@mDNA (IMB captured with mDNA), 10 μL of MB (4000 nm), IMB, and IMB@mDNA (each 10 mg/mL) was washed three times and diluted in 1000 μL of phosphate-buffered saline (PBS) and ultrasound for 10 min, respectively. Scanning electron microscopy (SEM) images of MB and IMB were performed with a Phenom ProX Desktop scanning electron microscope (Phenom, Netherlands) at an accelerating voltage of 4.8 KV. Of diluted samples, 2.5 μL was dropped on the silicon wafer and dried at 37 °C.
Cell culturing, plasma samples, and DNA extraction
Lovo cells and CCD-18Co cells were cultured in 25-cm2 tissue culture flasks in Roswell Park Memorial Institute (RPMI) 1640 medium. SW 480 cells and HCT 116 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM). All media were supplemented with 10% fetal bovine serum (FBS) (v/v) and 1% penicillin/streptomycin. The culture medium was replaced every other day and cultured until cells approach 80–90% confluence. Clinical plasma samples of 20 CRC patients and 20 healthy controls were obtained from the central laboratory of the Yangpu Hospital with informed consent (Shanghai, China). Medical Research Ethics Committee of Yangpu Hospital approved all experimental protocols. Plasma was collected in a 1.5-mL tube and stored at −80 °C. DNA samples from these cells and clinical plasma were extracted by DNA extraction kit (TIANGEN, China) according to the manufacturer’s instruction, described in the electronic supplementary material. The extracted samples were quantified using the SMA4000 UV–Vis Spectrophotometer and used immediately or stored in −20 °C.
Preparation of IMB
The preparation of IMB was processed according to the manufacturer’s standard protocol with some modifications. Ten microliters of 10 mg/mL MB coated with carboxyl groups was washed twice with 200 μL of 25-mM MES (pH = 6.0) with good mixing at room temperature and separated from the solution with a magnet strand. Fifty-five microliters of 25-mM MES and 5 μL of 0.2 mg/mL anti-5-mC antibody were mixed with the washed beads and incubated with slow rotation at room temperature for 30 min. Then 30 μL of 10 mg/mL fresh prepared EDC (in cold 25-mM MES) was added to the beads, and 10 μL of 25 mM MES was added to a final volume of 100 μL. The whole mixture was incubated for 2 h at room temperature with slow tilt rotation. After that, the IMB (anti-5mC antibody modified MB) was washed three times with 300 μL 0.01 M PBS solution and finally resuspended in 100 μL of PBS with 0.1% Triton X-100. The prepared IMB was stored at 4 °C until use.
Enrichment of mDNA by IMB
For specific mDNA isolation, 50 μL of 10 ng/μL DNA sample was incubated with 10 μL of 1 mg/mL IMB at room temperature for 30 min. Thereafter, three washing steps were carried out using 100 μL of 0.01 M PBS solution each time to ensure complete removal of other non-specifically adsorbed nucleic acids. Then, the methylated DNA isolated by IMB was eluted by a 50-μL TE buffer containing 5 μL 20 mg/mL proteinase K (50 °C, 10 min). Subsequently, proteinase K was inactivated at 95 °C for 10 min. The supernatant of mDNA was collected and quantified using the SMA4000 UV–Vis Spectrophotometer for amplification experiments.
Primer design
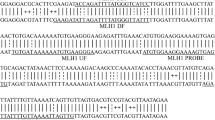
The H. sapiens Septin9 (NCBI Reference Sequence: NR_011683.1) was used as the target gene in this study. Plasmid containing an 851-bp fragment of wild-type Septin9 gene was constructed and selected to design 5 sets of LAMP primers by primer explorer V5 on the website (http://primerexplorer.jp/e/index.html, Eiken Chemicals Corporation, Tokyo, Japan). PCR primers targeting Septin9 were designed on the NCBI website (https://www.ncbi.nlm.nih.gov/). These specific primers are listed in Table S1. For verification of specificity, the designed primers and target sequences have been analyzed by the NCBI BLAST tool with the use of the nucleotide database as the reference. LAMP primers for β-actin genes were referred to a previous report (Table S1) [37].
LAMP assay and amplification analysis
For LAMP reaction, the whole reaction was carried out in 12.5-μL mixture containing 1 × ThermoPol Buffer (20-mM Tris-HCl, 10-mM (NH4)2SO4, 150-mM KCl, 2-mM MgSO4, 0.1% Tween 20), 6-mM MgSO4, 1-M betaine, 1.4-mM dNTP mixture, 1.6-μM FIP/BIP primer, 0.2-μM F3/B3 primer, 8-U Bst DNA polymerase, and 2.5-μL DNA sample with different concentrations. For real-time fluorescent LAMP, SYTO-9 was used as the fluorescent signal molecule in this study with an excitation at 470 nm and fluorescence emission at 525 nm, and 500-nM SYTO-9 was pre-added to the mixture. As for visualized detection, 100-μM neutral red was added before the amplification. The LAMP solution was amplified for 60 min at 65 °C on a LineGene 9640 real-time PCR system. Immediately following LAMP, a melting curve was performed by a dissociation analysis at 95–65 °C through detecting the change of fluorescence intensity. Threshold time (Tt) values were calculated using the built-in second-derivative algorithm from the LightCycler 480 software.
PCR assay
To further assess the accuracy of this developed method, clinical plasma samples were also tested by the traditional PCR method. MDNA from these samples were obtained by IMB enrichment according to the aforementioned enrichment procedure. According to the instructions of the PCR kit (Hieff® qPCR SYBR Green Master Mix from Yeasen Biotechnology (Shanghai, China)), each PCR reaction was carried in a 20-μL system composing of 0.2-μM forward primer (FP), 0.2-μM reverse primer (RP), 10-μL PCR reaction buffer, and 2-μL extracted sample. PCR was conducted on a LineGene 9640 real-time PCR system with a temperature profile of 95 °C for 5 min, followed by 40 cycles of amplification (95 °C for 10 s, 60 °C for 30 s).
Gel electrophoresis
The amplification products were analyzed using gel electrophoresis (Tanon EPS 300, Shanghai, China) to confirm the presence and size distribution of the amplicons. Ten microliters of amplified products mixed with 2 μL of loading buffer was resolved in 2% agarose gel using 1 × TAE buffer at 110 V for 30 ~ 40 min, with 5-μL YeaRed DNA nucleic acid gel staining. Five-microliter DNA Ladder (50 ~ 500 bp) was mixed with 2 μL of loading buffer and used to confirm product size. The gels were imaged by the imaging system (Tanon-4100).
Results and discussion
Strategy of fluorescent and colorimetric LAMP for mDNA detection
This mDNA detection platform composed isolation and enrichment of mDNA using anti-5mC antibody coated MB and a dual-modality (fluorescent and colorimetric) LAMP strategy (Scheme 1). Briefly, the total DNA was extracted from samples (e.g., cells, blood) using lysis buffer and incubated with IMB for 30 min, and mDNA was isolated due to the immunoaffinity between mDNA and anti-5mC antibody on the surface of IMB. After thoroughly washed three times to remove non-specifically adsorbed other nucleic acids and impurities, the captured mDNA was eluted by TE buffer (containing proteinase K to digest antibody, thereby improving the elution efficiency), and LAMP signal readout of eluted mDNA template was identified by both real-time fluorescence monitoring and directly naked-eye observation in one step. A pH indicator (neutral red, range 6.8–8.0) was used as for colorimetric detection [24]. As the LAMP reaction proceeds, the pH of the reaction solution reduced from 8.8 to less than 8.0 due to the continuously produced hydrogen ions (by-products of LAMP reaction), leading to a visible color change from yellow to pink, therefore making it a promising method for the application in clinical diagnostics owning to the portability, low cost, the independent of complicated equipment, and visualized detection. In addition, IMB enrichment provides the advantage of independence on bisulfite conversion and enzyme digestion as well as simplifying and shortening operation; the dual-modality analysis enables real-time monitoring and visualization with naked eyes, offering convenient, accurate, and quantitative detection.

Scheme of fast IMB enrichment-assisted dual-modality LAMP detection of specific methylated DNA target
Characterization of IMB
The morphology of MB and IMB was characterized by SEM. As shown in Fig. 1A, MB were monodispersed in water with an average diameter of 4000 nm. IMB were slightly different from pure MB due to the anti-5mC antibody conjugated on the surfaces of MB (Fig. 1B). The zeta potential of MB was 0.70±0.28 mV (Fig. 1C). After being modified with anti-5mC antibody, the zeta potential of IMB decreased to −0.47± 0.05 mV (Fig. 1C), demonstrating the successful conjugation of antibody on the surface of MB. When IMB was incubated with DNA sample, the zeta potential of IMB@mDNA increased to −0.15± 0.08 mV (Fig. 1C), indicating the mDNA were successfully enriched by IMB.

SEM images of A MB and B IMB. C Zeta potential of MB, IMB, and IMB@mDNA. Scale bar, 5 μm. Error bars represent the standard deviation from the mean based on three independent experiments. Number of experiments n = 3
Primer screening
Septin9 is a tumor suppressor gene; the hypermethylation of CpG islands in the promoter of Septin9 can repress the promoter activity and lose its tumor suppressor function [38]. Consequently, hypermethylated Septin9 serves as a biomarker for early cancer detection and monitoring treatment [39,40,41], which has been approved by the US FDA for colorectal cancer (CRC) screening [42]. Here, five pairs of LAMP primers (1, 2, 3, 4, 5) were designed according to the Septin9 gene (Table S1). Real-time fluorescence LAMP was developed for screening reliable primers to specifically target Septin9 using CRC cells (HCT 116) and normal colon cells (CCD-18Co) as positive control and negative control, respectively. In the presence of target, primer sets 2, 3, 4, and 5 could successfully trigger LAMP reaction before 20 min with high amplification efficiency (Fig. 2A), while primer set 1 only generated weak fluorescence signal in about 50 min. In the absence of target, primer sets 1 and 5 displayed the accurate negative signal; however, primer sets 2, 3, and 4 triggered either weak or strong false-positive signal unfortunately (Fig. 2B). As a result, primer set 5 was the most suitable primer pair for distinguishing positive samples from negative samples fast and accurately; thereby, primer set 5 was used for methylated Septin9 detection in the subsequent assay.

LAMP primers screening for detecting methylated Septin9 gene. A Real-time fluorescence monitoring LAMP reactions for positive control (HCT 116 cell as the target, mDNA 5 ng/uL) triggered by five primer sets. B Real-time fluorescence monitoring LAMP reactions for negative control (CCD-18Co cell as the blank, mDNA 5 ng/uL) triggered by five primer sets. The fluorescence of SYTO-9 was used as a fluorescence signal. LAMP condition: 65 °C, 1 h. Number of experiments n = 3
Sensitivity of fluorescent and colorimetric LAMP for mDNA detection
To achieve the optimal performance of IMB enrichment-assisted LAMP, the experimental conditions—size of MB (4000 nm), the ratio of anti-5mC antibody and MB (5 μg/mg), and incubation time for isolating mDNA (30 min)—were optimized (Fig. S1, see detailed results and discussion in the electronic supplementary material).
The sensitivity of LAMP was investigated by detecting methylated Septin9 using the three types of CRC cells (HCT 116 cells, SW480 cells, and Lovo cells) as targets. On the basis of the aforementioned optimization, mDNA was isolated through the specific immunoaffinity with IMB and methylated Septin9 was detected by the developed dual-modality LAMP. The fluorescence amplification signals were generated from 15 to 35 min or so by varying the concentration of mDNA from HCT 116 cells, SW480 cells, and Lovo cells (Fig. 3A, B, and C, respectively). The amplification Tt values were in linear correlation to the log of the initial concentration of target, according to the criterion of the detection limit of nucleic acid amplification detection [43, 44]; the detection limit of this method was the lowest detectable concentration—0.02 ± 0.002 ng/μL (RSD: 9.75%), 0.1 ± 0.04 ng/μL (RSD: 10.68%), 0.1± 0.03 ng/μL (RSD: 3.48%), respectively (Fig. 3D, E, and F)—indicating a potential of this method in quantification detection through real-time fluorescence LAMP. According to the result, as few as 0.25 ± 0.025 ng of mDNA (0.02 ng/μL in LAMP system with a total volume of 12.5 μL) could be detected by LAMP owing to its high sensitivity, excellent amplification efficiency, and good anti-interference, and the detection limit is much lower than that of the recently reported electrochemical sensing strategy [33] and field-effect transistor [41]. The distinct colorimetric discrimination (Fig. 3G, H, and I) implies that the method is able to detect the low abundant mDNA target. Gel electrophoresis analysis of these LAMP products in Fig. 3J, K, and L confirmed the accurate amplification of Septin9. The performance of this platform is much simpler, faster, inexpensive, and easy-to-use compared to the existing methods such as MS [45, 46] and high-resolution melting analysis (HRM) [47].

Evaluation of the sensitivity of dual-modality LAMP for detecting methylated Septin9 using HCT 116 cells, SW 480 cells, and Lovo cells as target samples, respectively. Real-time fluorescence monitoring LAMP with different mDNA concentrations from A HCT 116 cells, B SW 480 cells, and C Lovo cells. The fluorescence of SYTO-9 was used as a fluorescence signal. The linear relationship between the threshold time (Tt) and the logarithm of target concentrations using D HCT 116 cells, E SW 480 cells, and F Lovo cells samples. The Tt value indicates how long it took to detect a real signal from the samples: lower Tt values indicate a higher concentration of initial target. No Tt values were obtained from the amplification of negative controls in any of the above experiments. G, H, and I Corresponding colorimetric results after LAMP reaction displayed in A, B, and C. J, K, and L Gel electrophoresis analysis of LAMP products from A, B, and C. LAMP condition: 65 °C, 1 h. NC: negative control, CCD-18Co cells. Error bars represent the standard deviation from the mean based on three independent experiments. Number of experiments n = 3
In order to simulate the real detection settings, the performance of fluorescent and colorimetric LAMP for methylated Septin9 detection was further evaluated by using the mixed cancer cells with non-cancer cells containing different ratios as targets. Each type of CRC cells (HCT 116 cells, SW480 cells, and Lovo cells) was mixed with the normal colon cells (CCD-18Co cells), respectively, to obtain a final percentage of these cancer cells ranging from 50 to 0%. Total DNA extracted from these mixed samples was diluted to 10 ng/μL. Similarly, mDNA were enriched by IMB and methylated Septin9 was detected by the dual-modality LAMP. Real-time fluorescence LAMP curves (Fig. 4A, B, and C) showed that methylated Septin9 was successfully detected in all mixed cell samples containing only 0.1% HCT 116 cells (RSD: 6.60%, Fig. 4D), SW480 cells (RSD: 0.72%, Fig. 4E), and Lovo cells (RSD: 10.74%, Fig.4F). The corresponding colorimetric LAMP in Fig. 4G, H, and I also proved these results, suggesting the feasibility, robustness, and sensitivity of the dual-modality LAMP, which has the potential for clinical application.

Evaluation of the sensitivity of dual-modality LAMP for detecting methylated Septin9 using the CRC cells mixed with normal colon cells (CCD-18Co cells) at different ratios as simulated samples. HCT 116 cells, SW 480 cells, and Lovo cells were mixed CCD-18Co cells at a series of ratios (50–0%), respectively. The total number of cells is about 50,000 for each level. Real-time fluorescence monitoring LAMP for detecting methylated Septin9 in A HCT 116 cells, B SW 480 cells, and C Lovo cells at different percentages. The linear relationship between the threshold time (Tt) and the logarithm of the percentage of D HCT 116 cells, E SW 480 cells, and F Lovo cells. No Tt values were obtained from the amplification of negative controls in any of the above experiments. G, H, and I Corresponding colorimetric results after LAMP reaction displayed in A, B, and C. LAMP condition: 65 °C, 1 h. NC: negative control, CCD-18Co cells. Error bars represent the standard deviation from the mean based on three independent experiments. Number of experiments n = 3
Application to clinical plasma samples
Plasmas taken from 20 CRC patients and 20 healthy controls were tested to evaluate the performance of dual-modality LAMP in real plasma samples. First, the feasibility of dual-modality LAMP assay for clinical detection was validated by targeting a housekeeping gene (β-actin) using total DNA extracted from clinical samples as template [37]. As shown in Fig. 4, β-actin genes from all CRC patients’ samples and all negative samples were successfully detected by both fluorescence LAMP detection (Fig. 5A and B) and colorimetric LAMP detection (Fig. 5C and D) at different amplification times, proving the potential of this assay for practical medical settings. Then, mDNA samples from these clinical samples were obtained using IMB enrichment and detected by dual-modality LAMP assay. Both fluorescence LAMP results (Fig. 6A and B) and the colorimetric results (Fig. 6C and D) demonstrated that methylated Septin9 was detected in CRC patients, while there was no methylated Septin9 in all healthy controls. Simultaneously, the accuracy of this result was validated and showed high consistency with the gold standard PCR method (Fig. S2).

Evaluation of dual-modality LAMP for detecting β-actin genes in clinical samples. Real-time fluorescence monitoring LAMP for plasma samples from A 20 CRC patients and B 20 healthy controls. Corresponding colorimetric results after LAMP reaction from C 20 CRC patients and D 20 healthy controls (C1 to C20 represent plasma samples from 1 to 20 CRC patients; H1 to H20 represent plasma samples from 1 to 20 healthy controls. NC: negative control, ultrapure water)

Evaluation of dual-modality LAMP for detecting methylated Septin9 gene in clinical samples. Real-time fluorescence monitoring LAMP for plasma samples from A 20 CRC patients and B 20 healthy controls. Corresponding colorimetric results after LAMP reaction from C 20 CRC patients and D 20 healthy controls (C1 to C20 represent plasma samples from 1 to 20 CRC patients; H1 to H20 represent plasma samples from 1 to 20 healthy controls. NC: ultrapure water)
Many nanomaterial-based methods have been developed for mDNA detection (Table S2). Gold nanoparticles (AuNPs)-based colorimetry provides a relatively simple strategy for global methylation detection, while it needs more than 1 day to obtain the detection results [48]. Quantum organic semiconductor (QOS)–based surface-enhanced Raman spectroscopy (SERS) requires substantial skills to prepare QOS and its stability and accuracy for clinical application remain to be assessed [49]. Carbon nanoparticle-based fluorescence method [50] and MB-based electrochemistry [34] offer high sensitivity but suffer from multiple operations as well as the limited throughput of testing samples. Compared to these current methods, IMB enrichment simplifies the assay’s procedure and the LAMP technique enables high-throughput detection, which makes it more favorable for clinical applications. In particular, the one-pot dual-modality LAMP allows determining multiple methylation genes by designing specific primers and provides two signal reporting strategies. Nevertheless, the performance of this method (e.g., sensitivity) is expected to be further improved by improving enrichment and amplification efficiency, which makes it possible to be used for the early diagnosis of cancer.
Conclusions
In this study, we proposed a dual-modality LAMP that successfully detected methylated Septin9 from CRC cells and plasma samples. Taking advantage of the intrinsic isolation capability MB, IMB was employed to separate and enrich mDNA with high efficiency and simplicity, which avoids complicated pretreatment steps. Two signal reporting strategies, fluorescence and colorimetric LAMP, were developed for methylated Septin9 detection with the detection limit of 0.02 ± 0.002 ng/μL (RSD: 9.75%) as well as 0.1% CRC cells (RSD: 6.60%). This method was demonstrated to be highly consistent with the PCR method when applied for clinical samples testing, showing significant potential in specific methylation analysis for clinical diagnosis. The capability of this method to early diagnosis of cancer needs to be further assessed and improved via improving enrichment and amplification efficiency to meet the requirements of clinical applications.
References
Bird AP (1986) CpG-rich islands and the function of DNA methylation. Nature 321(6067):209–213. https://doi.org/10.1038/321209a0
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9(6):465–476. https://doi.org/10.1038/nrg2341
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41(2):178–186. https://doi.org/10.1038/ng.298
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome — biological and translational implications. Nat Rev Cancer 11(10):726–734. https://doi.org/10.1038/nrc3130
Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark SJ (2006) Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat Genet 38(5):540–549. https://doi.org/10.1038/ng1781
Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee T-H (2010) DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc 132(6):1782–1783. https://doi.org/10.1021/ja910264z
Watson JA, Watson CJ, McCann A, Baugh J (2010) Epigenetics: the epicenter of the hypoxic response. Epigenetics 5(4):293–296. https://doi.org/10.4161/epi.5.4.11684
Hori Y, Otomura N, Nishida A, Nishiura M, Umeno M, Suetake I, Kikuchi K (2018) Synthetic-molecule/protein hybrid probe with fluorogenic switch for live-cell imaging of DNA methylation. J Am Chem Soc 140(5):1686–1690. https://doi.org/10.1021/jacs.7b09713
Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, Chakravarthy A, Shen SY, Bakouny Z, Boccardo F, Steinharter J, Bouchard G, Curran CR, Pan W, Baca SC, Seo J-H, Lee G-SM, Michaelson MD, Chang SL et al (2020) Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med 26(7):1041–1043. https://doi.org/10.1038/s41591-020-0933-1
Nassiri F, Chakravarthy A, Feng S, Shen SY, Nejad R, Zuccato JA, Voisin MR, Patil V, Horbinski C, Aldape K, Zadeh G, De Carvalho DD (2020) Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med 26(7):1044–1047. https://doi.org/10.1038/s41591-020-0932-2
Zhang P, Qiu T, Liu L, Lv F, Li Z, Ying J, Wang S (2020) Conjoint analysis of DNA methylation for tumor differentiation using cationic conjugated polymers. ACS Appl Bio Mat 3(5):2867–2872. https://doi.org/10.1021/acsabm.0c00047
Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T, Kis O, Zhao Z, Spreafico A, Medina TS, Wang Y, Roulois D, Ettayebi I, Chen Z, Chow S et al (2018) Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563(7732):579–583. https://doi.org/10.1038/s41586-018-0703-0
Xu R-h, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, Yi S, Shi W, Quan Q, Li K, Zheng L, Zhang H, Caughey Bennett A, Zhao Q, Hou J, Zhang R, Xu Y, Cai H, Li G et al (2017) Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 16(11):1155–1161. https://doi.org/10.1038/nmat4997
Bhattacharjee R, Moriam S, Umer M, Nguyen N-T, Shiddiky MJA (2018) DNA methylation detection: recent developments in bisulfite free electrochemical and optical approaches. Analyst 143(20):4802–4818. https://doi.org/10.1039/C8AN01348A
Sina AAI, Carrascosa LG, Trau M (2019) DNA methylation-based point-of-care cancer detection: challenges and possibilities. Trends Mol Med 25(11):955–966. https://doi.org/10.1016/j.molmed.2019.05.014
Zhang S, Huang J, Lu J, Liu M, Li Y, Fang L, Huang H, Huang J, Mo F, Zheng J (2019) A novel fluorescent biosensor based on dendritic DNA nanostructure in combination with ligase reaction for ultrasensitive detection of DNA methylation. J Nanobiotechnol 17(1):121. https://doi.org/10.1186/s12951-019-0552-5
Feng Q, Qin L, Wang M, Wang P (2020) Signal-on electrochemical detection of DNA methylation based on the target-induced conformational change of a DNA probe and exonuclease III-assisted target recycling. Biosens Bioelectron 149:111847. https://doi.org/10.1016/j.bios.2019.111847
Cui X, Cao L, Huang Y, Bai D, Huang S, Lin M, Yang Q, Lu TJ, Xu F, Li F (2018) In vitro diagnosis of DNA methylation biomarkers with digital PCR in breast tumors. Analyst 143(13):3011–3020. https://doi.org/10.1039/C8AN00205C
Cao A, C-y Z (2012) Sensitive and label-free DNA methylation detection by ligation-mediated hyperbranched rolling circle amplification. Anal Chem 84(14):6199–6205. https://doi.org/10.1021/ac301186j
Chen X, Huang J, Zhang S, Mo F, Su S, Li Y, Fang L, Deng J, Huang H, Luo Z, Zheng J (2019) Electrochemical biosensor for DNA methylation detection through hybridization chain-amplified reaction coupled with a tetrahedral DNA nanostructure. ACS Appl Mater Interfaces 11(4):3745–3752. https://doi.org/10.1021/acsami.8b20144
Chung MT, Kurabayashi K, Cai D (2019) Single-cell RT-LAMP mRNA detection by integrated droplet sorting and merging. Lab Chip 19(14):2425–2434. https://doi.org/10.1039/C9LC00161A
Varona M, Anderson JL (2019) Visual detection of single-nucleotide polymorphisms using molecular beacon loop-mediated isothermal amplification with centrifuge-free DNA extraction. Anal Chem 91(11):6991–6995. https://doi.org/10.1021/acs.analchem.9b01762
Ma Y-D, Chen Y-S, Lee G-B (2019) An integrated self-driven microfluidic device for rapid detection of the influenza a (H1N1) virus by reverse transcription loop-mediated isothermal amplification. Sensors Actuators B Chem 296:126647. https://doi.org/10.1016/j.snb.2019.126647
Ding S, Chen R, Chen G, Li M, Wang J, Zou J, Du F, Dong J, Cui X, Huang X, Deng Y, Tang Z (2019) One-step colorimetric genotyping of single nucleotide polymorphism using probe-enhanced loop-mediated isothermal amplification (PE-LAMP). Theranostics 9(13):3723–3731. https://doi.org/10.7150/thno.33980
Yee EH, Sikes HD (2020) Polymerization-based amplification for target-specific colorimetric detection of amplified mycobacterium tuberculosis DNA on cellulose. ACS Sens 5(2):308–312. https://doi.org/10.1021/acssensors.9b02424
Phillips EA, Moehling TJ, Bhadra S, Ellington AD, Linnes JC (2018) Strand displacement probes combined with isothermal nucleic acid amplification for instrument-free detection from complex samples. Anal Chem 90(11):6580–6586. https://doi.org/10.1021/acs.analchem.8b00269
Lee D, Shin Y, Chung S, Hwang KS, Yoon DS, Lee JH (2016) Simple and highly sensitive molecular diagnosis of Zika virus by lateral flow assays. Anal Chem 88(24):12272–12278. https://doi.org/10.1021/acs.analchem.6b03460
Lalli MA, Chen X, Langmade SJ, Fronick CC, Sawyer CS, Burcea LC, Fulton RS, Heinz M, Buchser WJ, Head RD, Mitra RD, Milbrandt J (2020) Rapid and extraction-free detection of SARS-CoV-2 from saliva with colorimetric LAMP. medRxiv:2020.2005.2007.20093542. https://doi.org/10.1101/2020.05.07.20093542
Dao Thi VL, Herbst K, Boerner K, Meurer M, Kremer LPM, Kirrmaier D, Freistaedter A, Papagiannidis D, Galmozzi C, Stanifer ML, Boulant S, Klein S, Chlanda P, Khalid D, Barreto Miranda I, Schnitzler P, Kräusslich H-G, Knop M, Anders S (2020) A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci Trans Med 12(556):eabc7075. https://doi.org/10.1126/scitranslmed.abc7075
Wu J, Wei X, Gan J, Huang L, Shen T, Lou J, Liu B, Zhang JXJ, Qian K (2016) Multifunctional magnetic particles for combined circulating tumor cells isolation and cellular metabolism detection. Adv Funct Mat 26(22):4016–4025. https://doi.org/10.1002/ADFM.201504184
Jeong S, Park J, Pathania D, Castro CM, Weissleder R, Lee H (2016) Integrated magneto-electrochemical sensor for exosome analysis. Acs nano 10(2):1802–1809. https://doi.org/10.1021/ACSNANO.5B07584
Lin Q, Huang Z, Ye X, Yang B, Fang X, Liu B, Chen H, Kong J (2021) Lab in a tube: isolation, extraction, and isothermal amplification detection of exosomal long noncoding RNA of gastric cancer. Talanta 225:122090. https://doi.org/10.1016/J.TALANTA.2021.122090
Povedano E, Ruiz-Valdepenas Montiel V, Gamella M, Pedrero M, Barderas R, Pelaez-Garcia A, Mendiola M, Hardisson D, Feliu J, Yanez-Sedeno P, Campuzano S, Pingarron JM (2020) Amperometric bioplatforms to detect regional DNA methylation with single-base sensitivity. Anal Chem 92(7):5604–5612. https://doi.org/10.1021/acs.analchem.0c00628
Povedano E, Valverde A, Montiel VR, Pedrero M, Yanez-Sedeno P, Barderas R, San Segundo-Acosta P, Pelaez-Garcia A, Mendiola M, Hardisson D, Campuzano S, Pingarron JM (2018) Rapid electrochemical assessment of tumor suppressor gene methylations in raw human serum and tumor cells and tissues using immunomagnetic beads and selective DNA hybridization. Angew Chem Int Ed Engl 57(27):8194–8198. https://doi.org/10.1002/anie.201804339
Povedano E, Montiel VR, Valverde A, Navarro-Villoslada F, Yanez-Sedeno P, Pedrero M, Montero-Calle A, Barderas R, Pelaez-Garcia A, Mendiola M, Hardisson D, Feliu J, Camps J, Rodriguez-Tomas E, Joven J, Arenas M, Campuzano S, Pingarron JM (2019) Versatile electroanalytical bioplatforms for simultaneous determination of cancer-related DNA 5-methyl- and 5-hydroxymethyl-cytosines at global and gene-specific levels in human serum and tissues. ACS Sens 4(1):227–234. https://doi.org/10.1021/acssensors.8b01339
Healey MJ, Rowe W, Siati S, Sivakumaran M, Platt M (2018) Rapid assessment of site specific DNA methylation through resistive pulse sensing. ACS Sens 3(3):655–660. https://doi.org/10.1021/acssensors.7b00935
Ye X, Fang X, Li X, Kong J (2018) Gold nanoparticle-mediated nucleic acid isothermal amplification with enhanced specificity. Anal Chim Acta 1043:150–157. https://doi.org/10.1016/j.aca.2018.09.016
Molnar B, Toth K, Bartak BK, Tulassay Z (2015) Plasma methylated septin 9: a colorectal cancer screening marker. Expert Rev Mol Diagn 15(2):171–184. https://doi.org/10.1586/14737159.2015.975212
Potter NT, Hurban P, White MN, Whitlock KD, Lofton-Day CE, Tetzner R, Koenig T, Quigley NB, Weiss G (2014) Validation of a real-time PCR-based qualitative assay for the detection of methylated SEPT9 DNA in human plasma. Clin Chem 60(9):1183–1191. https://doi.org/10.1373/clinchem.2013.221044
Song L, Li Y (2015) Chapter four - SEPT9: a specific circulating biomarker for colorectal cancer. In: Makowski GS (ed) advances in clinical chemistry, vol 72. Elsevier, pp 171-204. https://doi.org/10.1016/bs.acc.2015.07.004
Dong D, Zhang J, Zhang R, Li F, Li Y, Jia Y (2020) Multiprobe assay for clinical SEPT9 methylation based on the carbon dot-modified liquid-exfoliated graphene field effect transistor with a potential to present a methylation panorama. ACS Omega 5(26):16228–16237. https://doi.org/10.1021/acsomega.0c02022
Song L, Wang J, Wang H, Chen Y, Jia J, Guo S, Liu H, Peng X, Xiao W, Gong Y, Yang B, Lu Y, Li Y (2018) The quantitative profiling of blood mSEPT9 determines the detection performance on colorectal tumors. Epigenomics 10(12):1569–1583. https://doi.org/10.2217/epi-2017-0154
Zhao Y, Chen F, Qin J, Wei J, Wu W, Zhao Y (2018) Engineered Janus probes modulate nucleic acid amplification to expand the dynamic range for direct detection of viral genomes in one microliter crude serum samples. Chem Sci 9(2):392–397. https://doi.org/10.1039/C7SC03994H
Ye X, Li Y, Wang L, Fang X, Kong J (2018) A novel exonuclease-assisted isothermal nucleic acid amplification with ultrahigh specificity mediated by full-length Bst DNA polymerase. Chem Commun 54(75):10562–10565. https://doi.org/10.1039/C8CC04577A
Snyder CM, Alley WR Jr, Campos MI, Svoboda M, Goetz JA, Vasseur JA, Jacobson SC, Novotny MV (2016) Complementary glycomic analyses of sera derived from colorectal cancer patients by MALDI-TOF-MS and microchip electrophoresis. Anal Chem 88(19):9597–9605. https://doi.org/10.1021/acs.analchem.6b02310
Lin XC, Zhang T, Liu L, Tang H, Yu RQ, Jiang JH (2016) Mass spectrometry based ultrasensitive DNA methylation profiling using target fragmentation assay. Anal Chem 88(2):1083–1087. https://doi.org/10.1021/acs.analchem.5b04247
Rizzi G, Lee JR, Dahl C, Guldberg P, Dufva M, Wang SX, Hansen MF (2017) Simultaneous profiling of DNA mutation and methylation by melting analysis using magnetoresistive biosensor array. ACS Nano 11(9):8864–8870. https://doi.org/10.1021/acsnano.7b03053
Lin Y-Z, Chang P-L (2013) Colorimetric determination of DNA methylation based on the strength of the hydrophobic interactions between DNA and gold nanoparticles. Acs Appl Mat Interfaces 5(22):12045–12051. https://doi.org/10.1021/AM403863W
Ganesh S, Venkatakrishnan K, Tan B (2020) Quantum scale organic semiconductors for SERS detection of DNA methylation and gene expression. Nat Comm 11(1):1135. https://doi.org/10.1038/S41467-020-14774-3
Dadmehr M, Hosseini M, Hosseinkhani S, Ganjali MR, Khoobi M, Behzadi H, Hamedani M, Sheikhnejad R (2014) DNA methylation detection by a novel fluorimetric nanobiosensor for early cancer diagnosis. Biosens Bioelectron 60:35–44. https://doi.org/10.1016/J.BIOS.2014.03.033
Funding
This work was jointly supported by the Shanghai Science and Technology Innovation Action Plan (20392001900), Shanghai Natural Science Foundation (20ZR1403000), National Natural Science Foundation of China (21974028), and the National Key R&D Program of China (2017YFA0205100).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
Material and methods (materials and reagents, DNA extraction); results and discussion (optimization of the experimental conditions); Fig. S1, Optimization of the experiment conditions of IMB for methylation DNA enrichment; Fig. S2, PCR method for detecting methylated Septin9 gene in clinical samples; the designed LAMP primers targeting Septin9 (primer sets 1–5); and β-actin, PCR primers targeting Septin9 are listed in Table S1. An overview on recently reported nanomaterial-based methods for the determination of methylated DNA is shown in Table S2. (DOCX 944 kb)
Rights and permissions
About this article

Cite this article
Lin, Q., Fang, X., Chen, H. et al. Dual-modality loop-mediated isothermal amplification for pretreatment-free detection of Septin9 methylated DNA in colorectal cancer. Microchim Acta 188, 307 (2021). https://doi.org/10.1007/s00604-021-04979-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-021-04979-8