
Abstract
Aims
Prior exposure to insulin-induced hypoglycemia was shown to increase glial acetate metabolism (GAM) during subsequent exposure to hypoglycemia in diabetic individuals. However, it remained unclear whether this effect was dependent on the disease state or the antecedent cause of hypoglycemia. We aimed to establish whether exposure to fasting-induced hypoglycemia was sufficient to produce alterations in GAM in non-diabetic individuals.
Methods
GAM was measured via carbon-13 magnetic resonance spectroscopy during infusion of [1-13C] acetate before and after a 72-h fast in six metabolically healthy men. All participants were male, aged 18–40 years, with a Body Mass Index of 20.0–27.9 kg/m2, who consented to reside at Pennington Biomedical Research Center for 4 days. The main outcome measure was the percent enhancement of cerebral [1-13C] bicarbonate (the primary metabolic byproduct of glial oxidation of [1-13C] acetate). Continuous glucose monitoring was used to measure hypoglycemic episodes during the 72-h fast.
Results
As expected, 72 h of fasting significantly reduced blood glucose levels and resulted in a high frequency of hypoglycemic episodes. Steady-state GAM increased from 53.5 ± 3.7 to 61.9 ± 1.7% following the 72-h fast (p = 0.005). This increase correlated with greater duration of hypoglycemia experienced during the fast (r = 0.967). In addition, subjects with greater GAM at baseline experienced a greater increase in the duration of hypoglycemia experienced during the 72-h fast (r = 0.979).
Conclusions
GAM has potential as a biomarker for susceptibility to hypoglycemic episodes.
Trail registration
Clinicaltrials.gov ID: NCT02690168.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypoglycemia-associated autonomic failure (HAAF) is a serious condition characterized by drastically reduced neuroendocrine responses to hypoglycemia, and the loss of physiological symptoms of hypoglycemia, or hypoglycemia unawareness [1]. The development of HAAF can lead to ever-worsening and often life-threatening episodes of severe hypoglycemia [2]. HAAF is a significant impediment to the maintenance of healthy plasma glucose levels in both type 1 and 2 diabetes [3, 4].
Glial cells are gaining acceptance for their regulatory functions in a variety of neurological conditions [5], including abnormal hypoglycemic counter-regulation [6,7,8,9,10,11]. Furthermore, neurospectroscopy studies have demonstrated alterations in glial metabolism in persons with diabetes (PWD) [12] and PWD with HAAF [13]. These studies assessed glial metabolism using carbon-13 magnetic resonance spectroscopy (13C MRS) combined with the infusion of 13C acetate (a monocarboxylate preferentially metabolized by glial cells) [14, 15]. Using this technique, Mason et al. [12] determined that glial acetate metabolism (GAM) during a hypoglycemic clamp was greater in PWDs than it was in controls. This was reflected by an increase in both the cerebral transport and acetate oxidation in diabetic subjects during hypoglycemia, yet these differences could not be directly attributed to the diabetic state per se. This is because the PWDs in this study displayed significantly reduced plasma levels of counter-regulatory hormones during the hypoglycemic clamp, indicating a high prevalence of HAAF in these subjects.
In a subsequent investigation by Gulanski et al. [13] GAM was assessed in PWDs with and without HAAF during a hypoglycemic clamp. GAM was greater in PWDs with HAAF than in those without HAAF and the non-diabetic control groups [13]. GAM was not different between PWDs without HAAF and controls. The authors suggest that in response to recurrent exposure to low glucose availability, individuals with HAAF may adapt by increasing their utilization of alternative fuels, such as acetate. Together, these results suggest that prior exposure to insulin-induced hypoglycemia in the diabetic disease state is associated with elevated GAM, but that diabetes itself is not sufficient to increase GAM.
Although consensus exists that HAAF is associated with abnormal GAM, it is unclear if hypoglycemia, independent of diabetes, can alter GAM. Furthermore, it is not understood if alterations in GAM are driven by exposure to hypoglycemia directly, as opposed to indirectly through treatment-induced hyperinsulinemia (the underlying antecedent of HAAF). The current study in metabolically healthy male subjects was, therefore, undertaken to understand these issues by using prolonged fasting as an alternative antecedent to hypoglycemia. A 72-h fast is known to produce moderate-to-severe hypoglycemia [16, 17] as well as hypoinsulinemia [16, 18, 19]. We hypothesized that in non-diabetic subjects exposed to a 72-h fast, hypoglycemia will induce increased GAM and any observed increase in GAM will be related to the magnitude of the hypoglycemia experienced during the fast.
Materials and methods
Subjects
An a priori power analysis (with a mean and standard deviation of 5 and 2.3, respectively) previously reported in a similar study [13] estimated that a sample size of 6 would provide adequate statistical power (0.85) for our primary outcome variable. Due to known gender differences in the counter-regulatory response to hypoglycemia [18, 20, 21], study enrollment was limited to males 18–40 years old with a Body Mass Index (BMI) between 20.0 and 27.9 kg/m2, who were willing to reside on the inpatient unit for 4 days. Subjects were excluded for prior diagnosis of type 1 or 2 diabetes mellitus, fasting glucose ≥ 110 mg/dL, hyperketonuria (≥ 15 mg/dL), contraindication to MRI, current use of prescription medications, history of or current eating disorder, history of obsessive compulsive disorder, contraindication to prolonged fasting, and consumption of ≥ 10 alcoholic drinks per week.
Study design
The study was conducted over 6 months from 11/2016 to 04/2017. A graphical representation of the study design is presented in Figure S1. Recruited subjects were evaluated for eligibility at a single screening visit. Eligible subjects were then scheduled for a 4-day (~ 80 h) inpatient stay. Subjects arrived at the inpatient unit in the morning after a 10-h overnight fast (day 0) and weight, vital signs and body composition (Lunar iDXA whole-body scanner, General Electric; Milwaukee, WI, USA) were measured. GAM was then measured via 13C MRS, after which subjects were fed and began a ≈ 75-h fast when only water could be consumed ad libitum. Continuous glucose monitoring (CGM) was utilized for the duration of the 72-h fast and CGM glucose levels were recorded every 5 min. At 72 h, measurement of GAM was repeated.
Clinical chemistry
Plasma or serum levels of blood glucose (BG), beta-hydroxybutyrate (BHB), free fatty acids (FFA), glucagon, insulin, epinephrine and norepinephrine levels were measured by the Pennington Biomedical Clinical Chemistry Core. See supplementary materials for further details of the specific methods used.
GAM
Glial acetate metabolism was determined via MRS performed on a GE 3T Signa HDxt magnet (GE Healthcare, Milwaukee, WI, USA) with a single loop (8-cm diameter) radiofrequency (rf) coil (Doty Scientific Inc., Columbia, SC, USA). The 8-cm rf coil was tuned and matched to 13C resonance frequency at 3 T, and pulse sequence calibrations were implemented on a 2-mM sodium [1-13C] acetate phantom and a 1.76-mM sodium [1-13C]bicarbonate phantom. Each participant was positioned in a 1H-quadrature head coil with the 13C rf coil placed against the participant’s cranium. Scout images were acquired to localize and identify occipital lobe of the brain. Specifically, a high-resolution T1-weighted sequence was used to acquire 5-mm-thick slices in the transverse plane. Higher order shimming routine was used to improve the linewidths of spectral resonances. Baseline MRS data was acquired prior to the start of infusion with a free induction decay (FID) chemical shift imaging [22] sequence by implementing the following parameters: time-to-repetition (TR) of 4 s, a flip angle of 75°, a spectral width of 2.2 kHz, 1024 sampling points, number of excitation (NEX) of 8, and 296 total number of scans. At the start of [1-13C] acetate infusion, the total number of scans parameter was changed to 1808 to acquire a separate dynamic scan over a 2-h time course. For each participant, a 400-mM sodium [1-13C] acetate was infused at a rate of 6 mg/kg (body weight) for the first 5 min followed by 3 mg/kg (body weight) for 55 min. Venous blood samples were obtained at 10-min intervals during the 13C MRS procedures, starting 20 min prior to [1-13C] acetate infusion, and were used for plasma 12C and 13C acetate determination via gas chromatography–mass spectrometry [(GC–MS); (see supplementary material for details of GC–MS methods)] Plasma fraction enrichment of 13C acetate was determined by dividing the plasma concentration of 13C acetate by total plasma acetate concentration {[13C acetate]/([13C acetate] + [12C acetate])}.
All spectral data were post-processed in jMRUI version 5.2 [23] and quantitation of metabolite peaks was performed in the time domain with the AMARES (Advanced Method for Accurate, Robust, and Efficient Spectral fitting) algorithm [24]. FID data were apodized with 2 Hz, fast Fourier transformed, and manually phased (zero-order phase). Confirmed bicarbonate resonance peak data was transferred into Matlab (MathWorks, Natick, MA, USA), normalized with respect to the total sum and fitted to a mono-exponential equation as previously described by Bluml et al. [25] (see supplementary materials for details of equations).
CGM
A G4 Platinum CGM system (Dexcom, San Diego, CA, USA) was used per manufacturer’s instructions to measure interstitial glucose. The sensor was calibrated every 12 h via capillary BG measurements by finger stick using a HemoCue B-glucose analyzer (HemoCue, Brea, CA, USA). After completing the fast, CGM data was exported to Microsoft Excel to determine the mean, maximum, and minimum BG, as well as percent time spent ≤ 70 mg/dL and frequency of hypoglycemic episodes. A hypoglycemic episode was defined as three or more consecutive CGM glucose readings ≤ 70 mg/dL (Level 1) or ≤ 54 mg/dL (Level 2) [26].
Statistics
Results are reported as mean ± SE. All statistical analyses were performed using SAS Version 9.4 (SAS Institute, Cary, NC, USA) via a linear mixed effect model for repeated measures. The mixed effect model allowed all subjects to be included in the analyses irrespective of missing MRS data (see “GAM” for details) and models the correlation between the same subjects using a compound symmetric covariance matrix. Two sample t tests, based on estimates from the mixed model, were used to determine differences between day 0 and day 3 levels of plasma levels of metabolites and hormones, steady-state PE of 13C bicarbonate, and metabolic modeling parameters. Distributional assumptions of the residuals from these models were investigated using both visual inspection as well as Shapiro–Wilks tests. Pearson’s correlation was used to determine the association between time ≤ 70 and steady-state PE of bicarbonate on days 0 and 3. For all statistical analyses, p values of < 0.05 were considered statistically significant.
Results
Baseline characteristics and demographics of the subjects are presented in Table 1. Subjects experienced a statistically significant weight loss (3.7 ± 0.4%) following the 72-h fast. All subjects had normoglycemia (85.1 ± 2.8 mg/dL) on day 0 (following a 12-h fast). Blood glucose levels were significantly reduced on day 3 (following the 72-h fast), exhibiting a 16% decrease relative to values obtained on day 0. Serum concentrations of FFA, glucagon, and epinephrine were significantly increased on day 3 (Table 2). Serum norepinephrine and acetate levels, however, were not changed (Table 2). There was no evidence of hyperinsulinemia on day 0 and insulin significantly decreased in response to the 72-h fast with concentrations below the level of detection (2.0 µU/mL) for 5/6 subjects. Similarly, there was no evidence of ketosis on day 0; however, the 72-h fast significantly increased BHB measured in serum and urine (Table 2).
[1-13C] acetate infusion produced a rapid increase in the plasma fractional enrichment of [1-13C] acetate within 10 min. Mean plasma fractional enrichment of [1-13C] acetate did not differ between day 0 and day 3 (70.32 ± 0.02 and 69.26 ± 0.02%, respectively; p = 0.61), nor did plasma levels of [1-13C] acetate (data not shown). Prior to the infusion of [1-13C] acetate, no spectral peaks associated with acetate metabolism were detected (Figure S2, panel A). With the onset of the [1-13C] acetate infusion, a rapid increase in the cerebral PE of 13C bicarbonate was observed (Fig. 1a). This enhancement reflects the glial oxidation of our infusate via the TCA cycle. Additional peaks associated with [1-13C] acetate oxidation by glial cells, including [1-13C] glutamine, [5-13C] glutamine, [1-13C] glutamate, and [5-13C] glutamate, were also observed in our spectra (Figures S2B and S2C). Total PE of 13C bicarbonate approached steady-state within 60–70 min (Fig. 1a) and persisted in our spectra following the offset of the [1-13C] acetate infusion (Figures S2B and S2C). Figure 1b displays the average curve-fit for all days 0 and 3 scans, respectively, at approximately 15-min intervals. A clear increase in the PE of 13C bicarbonate is shown in the average day 3 curve-fit relative to day 0, with a statistically significant increase in PE of bicarbonate on day 3 versus day 0 curve fits (p < 0.0001). The curve-fitting procedure also yielded the cerebral metabolic rate constants CMRACE, VTCA, VACE, and CO2 from Eq. (2) as described in supplimentary materials. There were no significant differences in the average values of these rate constants for day 0 versus day 3 (Table S1). Mean steady-state PE of 13C bicarbonate measured from 64 to 96 min following the initiation of the [1-13C] acetate infusion also increased on day 3 relative to day 0 (61.9 ± 1.7 versus 53.5 ± 3.7; p = 0.005). The day 0 values of PE of bicarbonate displayed increased inter-subject variability relative to day 3 values. Day 0 values ranged from 40.6 to 61.4%, while day 3 values ranged from 57.9 to 66.0% (Fig. 1c). Furthermore, in the 4 subjects with complete data on day 0 and day 3, PE of 13C bicarbonate was increased on day 3 relative to day 0 (filled circles with connecting lines in Fig. 1c). Altogether, these data reflect an increase in the rate of glial metabolism of [1-13C] acetate on day 3 relative to day 0.

Glial acetate metabolism (GAM) is increased following a 72-h fast. GAM was measured via carbon 13 magnetic resonance spectroscopy during [1-13C] acetate infusion. a Time course of the percent enhancement (PE) of 13C bicarbonate (HCO3; the primary metabolite of [1-13C] acetate oxidation) following the start of infusion in a representative subject after 12 h of fasting (day 0) and 72 h of fasting (day 3). The solid line represents the best fit of a mono-exponential function modeling GAM. b Comparison of the average best fit across subjects via a linear mixed effects model demonstrates a clear increase from day 0 to 3 (p < 0.0001). c Average steady-state PE of HCO3 was increased on day 3 relative to day 0 (p = 0.0053). Furthermore, the PE of HCO3 was increased on day 3 relative to day 0 in all subjects who completed both scans (symbols with connecting lines)
CGM glucose values were not significantly different from BG values measured at identical time points (p = 0.1835; data not shown). CGM glucose levels dropped well below 80 mg/dL during the first 24 h of the fast. On day 2, glucose levels rebounded to approximately 90 mg/dL before falling again toward hypoglycemic levels (< 70 mg/dL) during the last 24 h (Fig. 2, top panel). The subjects’ mean CGM glucose level during the 72-h fast was well within the normal range (70–150 mg/dL). However, the mean minimum CGM reading was below 70 mg/dL (Table 3), and the subjects experienced considerable time below 70 mg/dL (Fig. 2). The number of subjects who experienced at least one episode of Level 1 hypoglycemia was five (83.3%), four (60%), and five (83.3%) during the first, second, and third days of fasting, respectively. The number of subjects who experienced at least one episode of Level 2 hypoglycemia was two (33.3%), zero (0%), and one (16.7%) during the first, second, and third days of fasting, respectively. The percent time spent in the hypoglycemic state (< 70 mg/dL) was positively associated with the PE of bicarbonate measured on day 0 (Pearson’s r2 = 0.96, p = 0.004; Fig. 3a). Furthermore, an increased exposure to hypoglycemia was positively associated with increased GAM at the end of the 72-h fast (Pearson’s r2 = 0.94, p = 0.007; Fig. 3b).

Continuous glucose monitoring (CGM) demonstrates that 72 h of fasting leads to frequent bouts of hypoglycemia. CGM was used to monitor blood glucose levels at 5-min intervals throughout a 72-h fast. The mean blood glucose for all six subjects is plotted in the top panel. The gray shading represents the 95% confidence limit and the dotted line represents the clinical threshold for Level 1 hypoglycemia, 70 mg/dL. The lower panel represents the percent of subjects which experienced a hypoglycemic episode during each hour of the 72-h fast. A subject was determined to have had a hypoglycemic episode during a given hour of the fast if a CGM reading of ≤ 70 mg/dL was maintained for three consecutive CGM readings (15 min)

Individual differences in glial acetate metabolism (GAM) are related to the duration of hypoglycemia experienced during a 72-h fast. GAM, as measured by the percent enhancement of 13C bicarbonate (PE of HCO3) following an acetate infusion, is both predictive of and responsive to subsequent fasting-induced hypoglycemia. a Relationship between glia acetate metabolism measured just prior to 72 h of fasting (day 0) and the subsequent exposure to hypoglycemia during the fast (percent time ≤ 70 mg/dL). A correlation analysis between these two variables demonstrated a robust positive relationship between GAM and the duration of hypoglycemia experienced during fasting (Pearson’s r2 = 0.96, p = 0.004). b GAM measured following a 72-h fast (day 3) increase in a dose-dependent manner based on the magnitude of exposure to hypoglycemia during the fast (Pearson’s r2 = 0.94, p = 0.007)
Discussion
Alterations in GAM are associated with a variety of clinical conditions in humans [27] including abnormal hypoglycemic counter-regulation [12, 13]. In this study, we used 13C MRS in non-diabetic subjects to assess whether GAM increased following a 72-h fast (a natural initiator of hypoglycemia). The major findings of this study were: (1) a 72-h fast in metabolically healthy men increases GAM, (2) increased exposure to hypoglycemia during the fast is associated with larger increases in GAM, and (3) increased GAM measured prior to the 72-h fast is correlated with increased susceptibility to hypoglycemia during the fast. The first two findings support our original hypothesis, while the third finding was unexpected and suggests that 13C MRS is a promising marker for hypoglycemia risk.
Hypoglycemia is a major complication of type 1 and 2 diabetes. Recurrent exposure to insulin-induced hypoglycemia can lead to the development of HAAF (a life-threatening metabolic disorder associated with blunted hypoglycemic counter-regulation). Previous studies demonstrated that HAAF is associated with increased GAM in PWDs during acute hypoglycemia [12, 13]. Our results advance these finding by demonstrating that exposure to hypoglycemia alone (independent of diabetes or exposure to hyperinsulinemia) is sufficient to increase glial metabolism. Furthermore, using continuous glucose monitoring to document the magnitude of hypoglycemic exposure, we demonstrated for the first time that exposure to hypoglycemia leads to a dose-dependent increase in GAM.
Our observation that GAM may determine susceptibility to fasting-induced hypoglycemia is also noteworthy in light of previous work. Gulanski et al. [13] showed that during a hypoglycemic clamp, GAM in both PWDs and non-diabetic subjects was inversely related to their neuroendocrine responses to the clamp. Thus, subjects with increased GAM displayed reduced hypoglycemic counter-regulation, thereby demonstrating increased susceptibility to hypoglycemia. In addition, our finding regarding to the metabolic rate constants associated cerebral acetate metabolism (Table S1) are consistent with those of Gulanski et al. [13], with the exception that Gulanski et al. [13] did observe a 50% increase in CMRACE. Altogether, our findings along with those of Gulanski et al. [13] suggest that increased GAM is a predictor of increased susceptibility to hypoglycemia independent of disease state, acute glycemic condition (hypoglycemic versus euglycemic), and the cause of the hypoglycemia (fasting or hyperinsulinemia). These data suggest that further investigations into the relationship between GAM and HAAF may provide key mechanistic and diagnostic insights which could lead to better treatment outcomes for PWD.
The clinical management of HAAF is severely hampered by the lack of non-invasive, objective biomarkers useful for definitive diagnosis and for ascertaining the severity of HAAF in PWD. Although our study was not performed in PWDs, our results are applicable, since the physiological response to fasting-induced hypoglycemia is identical to that of insulin-induced hypoglycemia [28], and exposure to fasting-induced hypoglycemia produces HAAF-like symptoms in humans [29, 30]. Our observation that GAM during euglycemia may predict subsequent susceptibility to hypoglycemia raises the possibility for targeted assessment of GAM to assess hypoglycemic risk in PWD.
Several limitations of our study are worth noting. Our study was adequately powered to detect our primary endpoint, change in PE of bicarbonate. Yet, the robustness of correlations between the PE of bicarbonate and hypoglycemia, as well as our CGM dataset, would be strengthened by an increased sample size. Although CGM is commonly used to clinically monitor hypoglycemia, CGM glucose levels are not as accurate as blood glucose measurements. This limitation was offset somewhat by our choice of the Dexcom G4 device, which has been shown to be significantly more accurate than comparable devices [31]. In addition, our use of [1-13C] acetate instead [2-13C] acetate precluded determination of brain 13C acetate concentrations due to the close spectral proximity of 1-13C acetate and 5-13C glutamine, a downstream metabolite of 1-13C acetate oxidation (Figure S2B).
Conclusion
Using a unique natural paradigm to induce hypoglycemia in healthy men, and a sophisticated 13C MRS protocol, our study significantly advances the scope and importance of the relationship between GAM and hypoglycemia. Increases in GAM are directly proportional to the extent of hypoglycemia exposure, in a dose-dependent manner, and higher levels of GAM under euglycemic conditions are associated with susceptibility to hypoglycemia. These two findings are very important clinically, i.e., GAM is possibly a novel biomarker of hypoglycemic risk and its measurement by 13C MRS has the potential to provide a much-needed diagnostic tool for the detection and treatment of HAAF.
References
Ritter S (2017) Monitoring and maintenance of brain glucose supply: importance of hindbrain catecholamine neurons in this multifaceted task. In: harris rbs (ed) appetite and food intake: central control, 2nd edn. CRC Press/Taylor & Francis, Boca Raton, pp 177–204
Cryer PE (2013) Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 369:362–372
Cryer PE (2008) The barrier of hypoglycemia in diabetes. Diabetes 57:3169–3176
Segel SA, Paramore DS, Cryer PE (2002) Hypoglycemia-associated autonomic failure in advanced type 2 diabetes. Diabetes 51:724–733
Miller DW, Cookson MR, Dickson DW (2004) Glial cell inclusions and the pathogenesis of neurodegenerative diseases. Neuron Glia Biol 1:13–21
Sanders NM, Dunn-Meynell AA et al (2004) Third ventricular alloxan reversibly impairs glucose counterregulatory responses. Diabetes 53:1230–1236
Marty N, Dallaporta M, Foretz M et al (2005) Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Investig 115:3545–3553
Hermann GE, Viard E, Rogers RC (2014) Hindbrain glucoprivation effects on gastric vagal reflex circuits and gastric motility in the rat are suppressed by the astrocyte inhibitor fluorocitrate. J Neurosci 34:10488–10496
McDougal DH, Viard E, Hermann GE et al (2013) Astrocytes in the hindbrain detect glucoprivation and regulate gastric motility. Auton Neurosci 175:61–69
Rogers RC, Ritter S, Hermann GE (2016) Hindbrain cytoglucopenia-induced increases in systemic blood glucose levels by 2-deoxyglucose depend on intact astrocytes and adenosine release. Am J Physiol Regul Integr Comp Physiol 310:R1102–R1108
Rogers RC, McDougal DH, Hermann GE (2017) Hindbrain astrocyte glucodetectors and counterregulation. In: Harris RBS (ed) Appetite and food intake: central control, 2nd edn. CRC Press/Taylor & Francis, Boca Raton, pp 205–228
Mason GF, Petersen KF, Lebon V et al (2006) Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes 55:929–934
Gulanski BI, De Feyter HM, Page KA et al (2013) Increased brain transport and metabolism of acetate in hypoglycemia unawareness. J Clin Endocrinol Metab 98:3811–3820
Deelchand DK, Shestov AA, Koski DM et al (2009) Acetate transport and utilization in the rat brain. J Neurochem 109(Suppl 1):46–54
Fonnum F, Johnsen A, Hassel B (1997) Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia 21:106–113
Bak AM, Møller AB, Vendelbo MH et al (2016) Differential regulation of lipid and protein metabolism in obese vs. lean subjects before and after a 72-h fast. Am J Physiol Endocrinol Metab 311:E224–E235
Ding XQ, Maudsley AA, Schweiger U et al (2017) Effects of a 72 hours fasting on brain metabolism in healthy women studied in vivo with magnetic resonance spectroscopic imaging. J Cereb Blood Flow Metab 38:469–478 (271678X17697721)
Hojlund K, Wildner-Christensen M, Eshoj O et al (2001) Reference intervals for glucose, beta-cell polypeptides, and counterregulatory factors during prolonged fasting. Am J Physiol Endocrinol Metab 280:E50–E58
Vella A, Service FJ, O’Brien PC (2003) Glucose counterregulatory hormones in the 72-hour fast. Endocr Pract 9:115–118
Diamond MP, Jones T, Caprio S et al (1993) Gender influences counterregulatory hormone responses to hypoglycemia. Metab Clin Exp 42:1568–1572
Amiel SA, Maran A, Powrie JK et al (1993) Gender differences in counterregulation to hypoglycaemia. Diabetologia 36:460–464
Skamarauskas JT, Oakley F, Smith FE et al (2014) Noninvasive in vivo magnetic resonance measures of glutathione synthesis in human and rat liver as an oxidative stress biomarker. Hepatology 59:2321–2330
Vanhamme L, van den Boogaart A, Van Huffel S (1997) Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson 129:35–43
Stefan D, Cesare FD, Andrasescu A et al (2009) Quantitation of magnetic resonance spectroscopy signals: the jMRUI software package. Meas Sci Technol 20:104035
Bluml S, Moreno-Torres A, Shic F et al (2002) Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed 15:1–5
International Hypoglycaemia Study Group (2017) Glucose concentrations of less than 3.0 mmol/L (54 mg/dL) should be reported in clinical trials: a joint position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 40:155–157
Ross B, Lin A, Harris K et al (2003) Clinical experience with 13C MRS in vivo. NMR Biomed 16:358–369
Boyle PJ, Shah SD, Cryer PE (1989) Insulin, glucagon, and catecholamines in prevention of hypoglycemia during fasting. Am J Physiol 256:E651–E661
Adamson U, Lins PE, Grill V (1989) Fasting for 72 h decreases the responses of counterregulatory hormones to insulin-induced hypoglycaemia in normal man. Scand J Clin Lab Investig 49:751–756
Drenick EJ, Alvarez LC, Tamasi GC et al (1972) Resistance to symptomatic insulin reactions after fasting. J Clin Investig 51:2757–2762
Kropff J, Bruttomesso D, Doll W et al (2015) Accuracy of two continuous glucose monitoring systems: a head-to-head comparison under clinical research centre and daily life conditions. Diabetes Obes Metab 17:343–349
Acknowledgements
This work was supported in part by 1 U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health, which funds the Louisiana Clinical and Translational Science Center and P30 DK072476, the Louisiana Nutrition Obesity Research Center. This work was also supported by the Pennington Biomedical Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank the staff of the PBRC Inpatient and Outpatient clinics for their invaluable support of this study, as well as Dr. Robbie Beyl for providing statistical analyses and consultation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
Ethical standard statement
All study procedures and literature were approved by the Institutional Review Board at Pennington Biomedical Research Center (PBRC). All subjects provided informed consent prior to enrollment in the study (NCT02690168). All study procedures and measurements, except plasma acetate assays (conducted at the University of Tennessee, Knoxville, TN, USA), were performed at PBRC, Baton Rouge, LA, USA.
Informed consent
Informed consent was obtained from all individual participants in the study.
Additional information
Managed by Massimo Federici.
Electronic supplementary material
Below is the link to the electronic supplementary material.
592_2018_1180_MOESM2_ESM.tif
Figure S1. Study design including screening and inpatient study visit, as well as all procedures and meals. Abbreviations: WGT (weight), MRS (magnetic resonance spectroscopy scan), AEs (adverse events), EOS (end of study) (TIF 626 KB)
592_2018_1180_MOESM3_ESM.tif
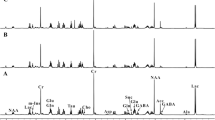
Figure S2. Representative spectra collected from the occipital/parietal cortex using carbon-13 magnetic resonance spectroscopy coupled with a 60-minute intravenous infusion of 1-13C acetate. Spectra were collected 20 minute prior to, and 120 minute following the start of the infusion. Each spectrum displayed represents the average data from a ≈20 minute time block. Spectral peaks corresponding to 1-13C acetate (Ac1), as well as the following downstream metabolites of 1-13C acetate oxidation are annotated: Gln5 (5-13C glutamine), Glu5 (5-13C glutamate), Gln1 (1-13C glutamine), Glu1 (1-13C glutamate), and HCO3 (1-13C bicarbonate). No natural abundance peaks corresponding to either acetate or its metabolites were detected in baseline spectra collected prior to the initiation of acetate infusion (A). All these peaks were all readily apparent during the [1-13C] acetate infusion and continued to be present shortly after termination of the [1-13C] acetate infusion (B). Spectra collected 90 minutes following the initiation of the infusion often lacked peaks for less abundant metabolites of 1-13C acetate oxidation, such as Glu5. In contrast, the peak for the major metabolite of 1-13C acetate oxidation, HCO3, continued to be present for the entire 120 minutes following the commencement of the infusion (C) (TIF 2148 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McDougal, D.H., Darpolor, M.M., DuVall, M.A. et al. Glial acetate metabolism is increased following a 72-h fast in metabolically healthy men and correlates with susceptibility to hypoglycemia. Acta Diabetol 55, 1029–1036 (2018). https://doi.org/10.1007/s00592-018-1180-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00592-018-1180-5