
Abstract
Purpose
Several recent studies have reported a possible association between gut microbiota and intervertebral disc degeneration; however, no studies have shown a causal relationship between gut microbiota and disc degeneration. This study was dedicated to investigate the causal relationship between the gut microbiota and intervertebral disc degeneration and the presence of potentially bacterial traits using two-sample Mendelian randomization.
Methods
A two-sample Mendelian randomization study was performed using the summary statistics of the gut microbiota from the largest available genome-wide association study meta-analysis conducted by the MiBioGen consortium. Summary statistics of intervertebral disc degeneration were obtained from the FinnGen consortium R8 release data. Five basic methods and MR-PRESSO were used to examine causal associations. The results of the study were used to examine the causal association between gut microbiota and intervertebral disc degeneration. Cochran's Q statistics were used to quantify the heterogeneity of instrumental variables.
Results
By using Mendelian randomization analysis, 10 bacterial traits potentially associated with intervertebral disc degeneration were identified: genus Eubacterium coprostanoligenes group, genus Lachnoclostridium, unknown genus id.2755, genus Marvinbryantia, genus Ruminococcaceae UCG003, family Rhodospirillaceae, unknown genus id.959, order Rhodospirillales, genus Lachnospiraceae NK4A136 grou, genus Eubacterium brachy group.
Conclusion
This Mendelian Randomization study found a causal effect between 10 gut microbiota and intervertebral disc degeneration, and we summarize the possible mechanisms of action in the context of existing studies. However, additional research is essential to fully understand the contribution of genetic factors to the dynamics of gut microbiota and its impact on disc degeneration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Intervertebral disc degeneration (IVDD) is a complex multifactorial pathophysiological process [1]. In this process, the destruction of the normal structure of the intervertebral disc (IVD) can often lead to a variety of spinal disorders, mainly characterized by low back pain, ultimately leading to loss of labor and a significant socio-economic burden [2]. Although the mechanisms underlying the development of IVD are currently unknown, there is evidence that microorganisms play an essential role in the development of IVDD [3,4,5]. Since Stirling et al. reported the relationship between Acinetobacter propionic and intervertebral discs in 2001 [6], research on the relationship between microorganisms and IVDD has been gradually enriched, especially in recent years, with the introduction of 16S rRNA amplicon sequencing (16S rRNA) and next-generation sequencing (NGS) as representative assay technologies [7,8,9]. The role of the microbiome in the development of IVDD in future studies could turn out to be striking for researchers [10].
Among these studies, we note that the relationship between the gut microbiota and IVDD has received a great deal of attention in recent years, and the existence of a "Gut-disc axis" has been envisaged for this purpose [10]. In the current research context, the mechanisms by which the gut microbiota contributes to the development of IVDD are unclear, and it is unclear whether the gut microbiota plays a positive or negative role in the development of IVDD. Therefore, a study is urgently needed to demonstrate the causal relationship between intestinal flora and IVDD.
Mendelian randomization (MR) is a method used to reveal potential causal relationships between exposure and outcome [11]. It can be characterized by its ability to use exposure-related genetic variation as a proxy for exposure to assess the association between exposure and outcome [12]. Previous publications have raised questions about the fundamental assumption that genetic analysis is the driver of the microbiome [13]. However, subsequent studies have countered this assertion from multiple perspectives by demonstrating that genetic factors do indeed play a role in shaping the variability of the gut microbiota [14,15,16]. In recent studies, MR has been widely used to explore the gut microbiota and various diseases, such as Autoimmune Diseases [17], major depressive disorder [18], and SLE [19]. In this study, using the genome-wide association study (GWAS) summary statistics from the MiBioGen (https://mibiogen.gcc.rug.nl)and FinnGen (https://www.finngen.fi) consortiums, a two-sample MR analysis was conducted to evaluate the causal association between gut microbiota and IVDD.
Methods
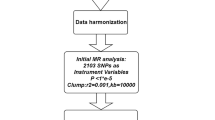
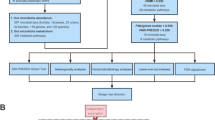
MR rests on three basic assumptions: (1) genetic variants are associated with the risk factor (relevance assumption), (2) those genetic variants are not associated with any known or unknown confounders (independence assumption), and (3) genetic variants affect the outcome only through the risk factor (exclusion restriction assumption) [12]. Based on the three basic assumptions above, we designed a study [20] (Fig. 1A) and developed a roadmap (Fig. 1B).

A Design ideas for the study. We designed this study based on three basic assumptions of MR. B The roadmap we developed based on our design
Data sources
In the conduct of our MR analysis, the exposure data were sourced from the MiBioGen consortium, which offers a comprehensive array of 211 bacterial traits. This dataset provides an extensive resource for identifying genetic associations with bacterial traits, serving as a pivotal element in establishing the instrumental variables necessary for our analysis [21]. The outcome data were drawn from the FinnGen Consortium's R8 release, a rich genomic resource characterized by extensive phenotypic information. Specifically, we extracted data corresponding to the phenotype labeled "Other intervertebral disc disorders" within this release. The careful selection of this label was imperative to ensure a precise and relevant definition of IVDD for our investigation [22].The choice of MiBioGen for exposure data and FinnGen for outcome data was driven by the comprehensive nature and high-resolution detail offered by both datasets, thereby enhancing the potential for accurate causal inference in our MR study. The alignment of these datasets allowed for a robust assessment of the genetic determinants of bacterial traits and their causal associations with IVDD.
Summary statistics for the gut microbial taxa were obtained from a large-scale multi-ethnic GWAS meta-analysis that included 18,340 individuals from 24 cohorts. Microbial composition was profiled by targeting three distinct variable regions of the 16S rRNA gene [21]. To account for differences in the sequencing depth, all datasets were rarefied to 10,000 reads per sample. Taxonomic classification was performed by direct taxonomic binning. In each cohort, only the taxa present in > 10% of the samples were included to explore the effect of host genetics on the abundance of gut bacterial taxa. The study-wide cutoffs included an effective sample size of at least 3000 individuals and their presence in at least three cohorts. A total of 211 taxa (131 genera, 35 families, 20 orders, 16 classes, and nine phyla) were included. After adjustment for age, sex, technical covariates, and genetic principal components, Spearman’s correlation analysis was performed to identify the genetic loci that affected the covariate-adjusted abundance of bacterial taxa.
GWAS summary statistics for IVDD were obtained from the FinnGen Consortium R8 release data [23]. The “Other intervertebral disc disorders” phenotype was adopted in the current study. This GWAS included 33,360 cases and 248,831 controls [22]. We defined this phenotype based on the International Classification of Diseases, 10th Revision (ICD-10), which is widely used in previous studies [24,25,26]. Diseases have been classified according to the ICD-10 coding system within FinnGen's phenotypic library, thus ensuring the reliability of utilizing FinnGen for research purposes [27].
No ethical approval was necessary for the publicly available de-identified data.
Instrumental variable (IV)
The following selection criteria were used to select the IVs: (1) single nucleotide polymorphisms (SNPs) associated with each genus at the locus-wide significance threshold (P < 1.0 × 10–5) were selected as potential IVs [12]; (2) 1000 Genomes Project European sample data were used as the reference panel to calculate the linkage disequilibrium (LD) between the SNPs, and among those SNPs that had R2 < 0.001 (clumping window size = 10,000 kb), only the SNPs with the lowest p values were retained; (3) SNPs with minor allele frequency (MAF) ≤ 0.01 were removed; and (4) when palindromic SNPs existed, the forward strand alleles were inferred using allele frequency information.
We employed a stringent criterion for the selection of IVs, which necessitated the identification of SNPs significantly associated with the bacterial traits of interest. This is crucial as the strength and validity of the MR analysis hinge on the quality of the IVs. During the IV selection process, we discovered that a subset of bacterial traits did not have any SNPs that met our eligibility requirements. Specifically, 5 out of the 211 bacterial traits were identified as having zero eligible SNPs, indicating a lack of suitable IVs for these traits.
Acknowledging the importance of robust IVs in MR analysis, we made the decision to exclude these five bacterial traits from the subsequent stages of our analysis. The absence of appropriate IVs for these traits could undermine the validity of any causal inferences drawn. Consequently, the refined analysis proceeded with the remaining 206 bacterial traits, each backed by appropriate IVs, thereby maintaining the integrity of our MR study (Supplementary Table 6).
Mendelian randomization analysis
To investigate the causal relationship between gut microbiota and IVDD, we performed an MR analysis [28]. Causal effects were calculated by dividing the SNP-outcome effect by the SNP-exposure effect estimate [11, 29]. To contain multiple SNPs, various tests were performed, including inverse variance weighted (IVW), weighted median and MR-Egger [20]. Cochrane's Q tests were performed to assess heterogeneity between SNPs associated with each classification [28]. In the presence of heterogeneity (p < 0.05), a random-effects IVW test was used to provide a more conservative but robust estimate [12]. The weighted median test produced consistent estimates when ≥ 50% of the weights were from valid IVs. The MR-Egger regression test allowed for the presence of polymorphism in more than 50% of the IVs [30].
To elucidate the causal relationships between bacterial traits and IVDD, we applied a robust array of MR methods, each offering unique advantages and serving as mutual validation for the analyses conducted. These methods include Inverse Variance Weighted (IVW), Weighted Median, MR-Egger, Weighted Mode, and Simple Mode. Our primary method of analysis was the IVW approach, which is traditionally favored for its precision in basic causal estimation under the assumption that all SNPs employed are valid instrumental variables. The validity of the IVW results, and hence their credibility in causality assessment, hinges on this crucial assumption. To assess the validity of the SNPs used as instrumental variables and ensure they are not subject to heterogeneity, which could bias the estimates, we utilized Cochran’s Q statistic. The heterogeneity analysis indicated no significant heterogeneity among the SNPs for the identified bacterial traits (Q-value > 0.05, Table 1), suggesting that the IVW estimates are reliable. Additionally, we conducted MR-Egger regression to examine the presence of horizontal pleiotropy, where an instrumental variable affects the outcome through pathways other than the exposure of interest. Furthermore, the MR-PRESSO global test was employed to detect outliers, which can be indicative of SNPs that deviate significantly from the MR assumptions.
All statistical analyses were performed using R (version 4.2.2). IVW, weighted median and MR-Egger regression methods were performed using the "TwoSampleMR" package (version 0.5.6). The MR-PRESSO test was performed using the "MRPRESSO" package. The MR-PRESSO tests were performed using the "MRPRESSO" (version 1.0) package.
Results
Our analysis provided a robust examination of the causal relationships between specific bacterial traits and IVDD. Out of 211 bacterial traits, ten were identified with a significant causal link to IVDD, as established through an extensive IV analysis. Sensitivity assessments including MR-Egger regression and MR-PRESSO tests confirmed the absence of horizontal pleiotropy and heterogeneity, underscoring the precision of our findings. Visual representations such as scatter, funnel, and forest plots corroborated the positive or negative associations of these bacterial traits with IVDD, enhancing the interpretability of the genetic influence. The 'leave-one-out' sensitivity analysis further validated our results, demonstrating their consistency and resilience to the potential bias of individual SNPs. This thorough analytical approach affirms the reliability of our identified bacterial traits as potential contributors to IVDD, paving the way for targeted research.
All 211 bacterial traits were analyzed using MR and the 10 bacterial traits (IVW-p value < 0.05, Q-value > 0.05, p value > 0.05) that we associated with IVDD were: genus Eubacterium coprostanoligenes group, genus Lachnoclostridium, unknown genus id. 2755, genus Marvinbryantia, genus Ruminococcaceae UCG003, family Rhodospirillaceae, unknown genus id.959, order Rhodospirillales, genus Lachnospiraceae NK4A136 grou, genus Eubacterium brachy group (Table 1). The SNPs corresponding to each exposure factor, information corresponding to each SNP, and information on each SNP in the outcome can be found in Supplementary Table 2.
Within our MR analysis, the initial step involved conducting IV extraction on data associated with all 211 bacterial traits. During this process, it was observed that 5 of these bacterial traits lacked appropriate instrumental variables, as the count of eligible SNPs was 0. Following the exclusion of these five bacterial traits, the subsequent analysis was carried out on the remaining 206 bacterial traits (Supplementary Table 6).
Five methods, IVW, weighted median, MR-Egger, weighted mode, and simple mode, were used to analyze the data (Figs. 2, 3) (Supplementary Table 3). The IVW method is mainly used for basic causal estimation, which provides the most accurate results when all selected SNPs are valid IVs [31]. IVW results are generally considered to be the most credible when assessing causality [27]. Following sequential analysis using MR on 211 bacterial traits, a subset of 10 bacterial traits was identified as having a statistically significant causal association with IVDD, as determined by screening the IVW-p value (Table 1). Cochran’s Q statistic was used to quantify the heterogeneity among the selected SNPs, and the results showed that there was no evidence (Q-value > 0.05) of heterogeneity (Table 1). The horizontal pleiotropy between IVs and outcomes was assessed by MR-Egger regression, and the results showed that there was no evidence (p value > 0.05) of horizontal pleiotropy (Table 1). MR-PRESSO was also used to test for Outlier and during the test we found p value < 0.05 for genus Marvinbryantia (Table 2), so we did further testing and the results confirmed that there was no Outlier in the data [32,33,34,35,36].

MR analysis for 10 bacterial traits using five methods: inverse variance weighted, weighted median, MR-Egger, weighted mode, and simple mode. We plotted the primary outcome for 10 bacterial traits in a scatterplot, where each point represents an instrumental variable (IV), and the error bars represent 95% confidence intervals. The x-axis shows the effect of each IV on exposure, and the y-axis shows the effect of the IV on the outcome. The colored lines show the fitted results, and the slope of the line reflects the causal effect of exposure on the outcome

Plot of leave-one-out method, which involves removing each SNP one by one, calculating the meta-effect of the remaining SNPs, and observing whether the results change after removing each SNP. If the results change significantly after removing one SNP, it means that one SNP has a large influence on the results, which is undesirable. Ideally, the results will not change much after removing each SNP one by one. In our analysis, the results did not vary much along the overall error line after removing each SNP, which indicates that the results are robust
We plotted scatter plots of the primary outcomes for the 10 bacterial traits (Fig. 2), where each point represents an IV and the line on the points represent 95% confidence intervals. The horizontal coordinates show the effect of each IV on exposure, and the vertical coordinates show the effect of the IV on outcome. The colored lines indicate the fitting results, and the slope of the lines reflects the effect of exposure on outcome. The plots suggest that the expression of genus Eubacterium brachy group and genus Marvinbryantia is positively associated with the incidence of IVDD. Similarly, genus Eubacterium coprostanoligenes group, genus Lachnoclostridium, unknown genus id. 2755, genus Ruminococcaceae UCG003, family Rhodospirillaceae, unknown genus id.959, order Rhodospirillales, genus Lachnospiraceae NK4A136 grou, is negatively associated with the incidence of IVDD.
We plotted a funnel plot of the main analyses of the 10 bacterial traits, where each point represents an IV, which shows a relatively symmetrical shape (Fig. 4). This indicates that our results have a small bias and that there is heterogeneity among SNPs.

Funnel plots of the 10 bacterial traits primarily analyzed, where each point represents an IV, and the shape of the funnel plots is relatively symmetrical
We mapped forest plots of the main analyses of 10 bacterial traits, where each point represents an IV and each horizontal solid line reflects the outcome estimated by a single SNP using the Wald ratio method (Fig. 3). Some solid lines are entirely to the left of 0, suggesting that an increase in the trait reduces the risk of IVDD; some solid lines are entirely to the right of 0, suggesting that an increase in the trait elevates the risk of IVDD; and some solid lines cross 0, indicating that the results are not significant. The results of a single SNP are not a substitute for exposure, and a reasonable result can only be obtained by looking at the combined effect, which is the bottom red line. This is consistent with the conclusions of the previous scatter plot.
The sensitivity analysis of MR analysis included the test of pleiotropy, the test of heterogeneity, and the leave-one-out method. For the test of pleiotropy, we used MR-PROSS to detect gene-level pleiotropy and confirmed the absence of horizontal pleiotropy when the p value was greater than 0.05. In our study species, all the results were greater than 0.05, indicating no pleiotropy (Table 2). For the test of heterogeneity, we used Cochran’s Q test to detect heterogeneity and confirmed the absence of heterogeneity when Q-value was greater than 0.05 (Table 2). The results of the study did not need to account for the effect of heterogeneity. The “leave-one-out” method involves removing each SNP step by step (Fig. 5), calculating the meta effect of the remaining SNPs, and observing whether the results change after removing each SNP. If the results change a lot after removing an SNP, it means that there is a SNP that has a great influence on the results, which is undesirable. Ideally, the results should not change much after removing each SNP step by step. In our analysis, the results did not change much at the overall error line after removing each SNP, which indicates the reliability of the results.

Forest plots of the 10 bacterial traits for the primary analysis, where each point represents an instrumental variable (IV) and each horizontal solid line reflects the Wald ratio estimate from a single SNP. Some of the solid lines are entirely to the left of 0, indicating that an increase in the trait decreases the risk of IVDD; some are entirely to the right of 0, indicating that an increase in the trait increases the risk of IVDD; and some intersect 0, indicating that the effect is not significant. The results of a single SNP are not a valid proxy for exposure, and a reasonable estimate can only be obtained by looking at the combined effect (i.e., the red line at the bottom)
Discussion
In this study, we performed MR analysis to determine the potential causal relationship between 211 bacterial traits of the gut microbiota and IVDD. This is a shred of positive evidence for the existence of the “Gut-disc axis”. Ultimately, we found a causal effect between 10 gut microbiota and IVDD, namely the genus Eubacterium coprostanoligenes group, genus Lachnoclostridium, and unknown genu id.2755. genus Marvinbryantia, genus Ruminococcaceae UCG003, family Rhodospirillaceae, unknown genus id.959, order Rhodospirillales, genus Lachnospiraceae NK4A136 group, genus Eubacterium brachy group.
According to our study, the genus Eubacterium coprostanoligenes was concluded to be a protective factor against IVDD. As a group of anaerobic gram-positive bacteria, the genus Eubacterium coprostanoligenes can convert cholesterol to unabsorbable coprostanol, thereby regulating cholesterol levels [37]. In a previous study, it was demonstrated that cholesterol promotes IVDD by mediating apoptosis and pyroptosis in NP cells and metabolism of ECM [38]. Additionally, cholesterol metabolites affect the immune microenvironment by enriching immunosuppressive cells, inhibiting immune effector cells, and inhibiting antigen presentation. Wang L et al. showed that during the development of IVDD, the immune microenvironment of the intervertebral disc changes, characterized by infiltration of CD68 macrophages, T cells (CD4 /CD8) and neutrophils [39], accompanied by the release of inflammatory factors that are thought to be promoters of pain and IVDD [38, 39]. Targeting cholesterol metabolism provides novel insights into IVDD. Therefore, Future studies should elucidate the exact relationship between the genus Eubacterium coprostanoligenes and cholesterol levels in IVDD.
Genus Lachnoclostridium is thought to be causally linked to a variety of diseases, but the exact mechanisms are not yet clearly described. It is known to be involved in the synthesis of glutamate, butyrate, serotonin, and GABA, and we consider that these mediators may be responsible for the influence of the genus Lachnoclostridium on the development of IVDD; however, researchers will need to further studies are required to determine the role of the genus Lachnoclostridium in the development of IVDD.
There is a correlation between the genus Marvinbryantia and low skeletal muscle mass, which may arise by mediating a reduction in butyrate [40]. Low skeletal muscle mass has been previously studied for its apparent correlation with IVDD, a peri-spinal change that predicts altered disc biomechanics and is recognized as an important factor in the initiation of IVDD [41,42,43].
The Genus Ruminococcaceae UCG003 belongs to the Ruminococcaceae family, and many bacteria in this family are known to be general butyrate-producing bacteria [44]. The effect of Ruminococcaceae UCG003 on IVDD may be mediated by butyrate, as shown by Jia et al. [45]. to alleviate IVDD through inflammatory responses and NF-κB activation in intervertebral disc tissue.
The genus Lachnospiraceae NK4A136, representing a type of butyrate-producing bacteria, has been found to maintain epithelial barrier integrity in mice and is negatively correlated with intestinal permeability [46]. The genus Lachnospiraceae NK4A136 group is thought to play a key role in maintaining gut barrier stability [47], and epithelial barrier integrity is thought to be one of the main mechanisms of action in the "Gut-disc axis" [10]. Therefore, we propose that the genus Lachnospiraceae NK4A136 group aims to slow down the development of IVDD by maintaining the integrity of the epithelial barrier.
In the available studies, descriptions of unknown genu id.2755, family Rhodospirillaceae, unknown genus id.959, order Rhodospirillales, and genus Eubacterium brachy group are rare; therefore, we were also unable to search for their potential pathways of action on IVDD in the available studies. This is worth exploring in future studies, which will need to rely on advances and developments in existing experimental techniques.
This study had several limitations. 1. When we obtained the GWAS data source for gut microbiota, we noted that it was still a relatively small sample size compared to the GWAS data for other factors, although it is already the largest currently available for gut microbiota. Our findings are potentially subject to future changes if GWAS data with larger sample sizes, stratified by more countries, regions, ethnicities, and so on, are available. 2. In studying bacterial traits, our study was limited to hierarchical traits (class, family, genus, order, and phylum), and we were not able to study the species; however, this limitation stems from the backwardness of the current assay technology and that, in the future, more advanced technologies may be available to explore the impact of the gut microbiota on IVDD at the species level. 3. In the screening step for IVs, we used P < 1.0 × 10–6 instead of the widely accepted P < 1.0 × 10–8 [48], to obtain more SNPs, which in turn reduced the reliability of the results to some extent. This reduces the reliability of the results to some extent; however, the results are still plausible [28, 30, 32, 49, 50]. 4. Rothschild et al. (2018) challenged the role of host genetics in shaping the gut microbiota [13]. However, subsequent studies have rebutted this claim and demonstrated that genetic factors also contribute to variation in the gut microbiota [21, 51, 52]. We adopted this perspective to investigate the influence of the gut microbiota on IVDD, and we believe our results are robust. Nevertheless, our conclusions require further validation by future studies.
In conclusion, this study provides robust support for the existence of a "gut-disc axis." However, further investigation is warranted to unravel the influence of genetic factors on the intricacies of the gut microbiota dynamics. Additionally, a deeper exploration is needed to understand how the gut microbiota's role varies under distinct bacterial trait classifications and its subsequent impact on disc degeneration. Moreover, this study highlights pivotal areas that merit consideration as potential avenues for future research. These include cholesterol dynamics, the nuances of the immune microenvironment, and the role of butyrate. These directions hold promise in shedding light on critical mechanisms underlying the interaction between gut microbiota and disc degeneration.
Data availability
The datasets analyzed in the current study are available from the MiBioGen (https://mibiogen.gcc.rug.nl) and the FinnGen repository (https://www.finngen.fi).
Code availability
The analyses involved in this study were all completed in R via the TwoSampleMR-package.
References
Balagué F, Mannion AF, Pellisé F et al (2012) Non-specific low back pain. Lancet (London, England) 379:482–491. https://doi.org/10.1016/s0140-6736(11)60610-7
Deyo RA, Weinstein JN (2001) Low back pain. N Engl J Med 344:363–370. https://doi.org/10.1056/nejm200102013440508
Rajasekaran S, Soundararajan DCR, Tangavel C et al (2020) Human intervertebral discs harbour a unique microbiome and dysbiosis determines health and disease. Eur Spine J 29:1621–1640. https://doi.org/10.1007/s00586-020-06446-z
Lin L, Luo P, Yang M et al (2022) Causal relationship between osteoporosis and osteoarthritis: A two-sample Mendelian randomized study. Front Endocrinol 13:1011246. https://doi.org/10.3389/fendo.2022.1011246
Yao B, Cai Y, Wang W et al (2023) The effect of gut microbiota on the progression of intervertebral disc degeneration. Orthop Surg 15:858–867. https://doi.org/10.1111/os.13626
Stirling A, Worthington T, Rafiq M et al (2001) Association between sciatica and Propionibacterium acnes. Lancet (London, England) 357:2024–2025. https://doi.org/10.1016/s0140-6736(00)05109-6
Shanmuganathan R, Tangavel C, Sri-Vijay-Ananad KS et al (2022) Comparative metagenomic analysis of human intervertebral disc nucleus pulposus and cartilaginous end plates. Front Cardiovasc Med 9:927652. https://doi.org/10.3389/fcvm.2022.927652
Fritzell P, Welinder-Olsson C, Jönsson B et al (2019) Bacteria: back pain, leg pain and Modic sign-a surgical multicentre comparative study. Eur Spine J 28:2981–2989. https://doi.org/10.1007/s00586-019-06164-1
Alamin TF, Munoz M, Zagel A et al (2017) Ribosomal PCR assay of excised intervertebral discs from patients undergoing single-level primary lumbar microdiscectomy. Eur Spine J 26:2038–2044. https://doi.org/10.1007/s00586-017-5141-4
Li W, Lai K, Chopra N et al (2022) Gut-disc axis: a cause of intervertebral disc degeneration and low back pain? Eur Spine J 31:917–925. https://doi.org/10.1007/s00586-022-07152-8
Sekula P, Del Greco MF, Pattaro C et al (2016) Mendelian randomization as an approach to assess causality using observational data. J Am Soc Nephrol 27:3253–3265. https://doi.org/10.1681/asn.2016010098
Emdin CA, Khera AV, Kathiresan S (2017) Mendelian Randomization. JAMA 318:1925–1926. https://doi.org/10.1001/jama.2017.17219
Rothschild D, Weissbrod O, Barkan E et al (2018) Environment dominates over host genetics in shaping human gut microbiota. Nature 555:210–215. https://doi.org/10.1038/nature25973
Fassarella M, Blaak EE, Penders J et al (2021) Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut 70:595–605. https://doi.org/10.1136/gutjnl-2020-321747
McConnell RB (1971) Genetics and gastroenterology. Gut 12:592–598. https://doi.org/10.1136/gut.12.7.592
Sanna S, Kurilshikov A, van der Graaf A et al (2022) Challenges and future directions for studying effects of host genetics on the gut microbiome. Nat Genet 54:100–106. https://doi.org/10.1038/s41588-021-00983-z
Xu Q, Ni JJ, Han BX et al (2021) Causal relationship between gut microbiota and autoimmune diseases: a two-sample Mendelian randomization study. Front Immunol 12:746998. https://doi.org/10.3389/fimmu.2021.746998
Chen M, Xie CR, Shi YZ et al (2022) Gut microbiota and major depressive disorder: a bidirectional Mendelian randomization. J Affect Disord 316:187–193. https://doi.org/10.1016/j.jad.2022.08.012
Xiang K, Wang P, Xu Z et al (2021) Causal effects of gut microbiome on systemic lupus erythematosus: a two-sample Mendelian randomization study. Front Immunol 12:667097. https://doi.org/10.3389/fimmu.2021.667097
Ference BA, Holmes MV, Smith GD (2021) Using Mendelian randomization to improve the design of randomized trials. In: Cold Spring Harbor perspectives in medicine. 2021; p. 11. https://doi.org/10.1101/cshperspect.a040980
Kurilshikov A, Medina-Gomez C, Bacigalupe R et al (2021) Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet 53:156–165. https://doi.org/10.1038/s41588-020-00763-1
Kurki MI, Karjalainen J, Palta P et al (2023) FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613:508–518. https://doi.org/10.1038/s41586-022-05473-8
Kurki MI, Karjalainen J, Palta P et al (2022) https://doi.org/10.1101/2022.03.03.22271360
Jin P, Xing Y, Xiao B et al (2023) Diabetes and intervertebral disc degeneration: a Mendelian randomization study. Front Endocrinol 14:1100874. https://doi.org/10.3389/fendo.2023.1100874
Ou-Yang DC, Kleck CJ, Ackert-Bicknell CL (2023) Genetics of intervertebral disc degeneration. Curr Osteoporos Rep 21:56–64. https://doi.org/10.1007/s11914-022-00769-0
Zhou J, Mi J, Peng Y et al (2021) Causal associations of obesity with the intervertebral degeneration, low back pain, and sciatica: a two-sample mendelian randomization study. Front Endocrinol 12:740200. https://doi.org/10.3389/fendo.2021.740200
Pagoni P, Dimou NL, Murphy N et al (2019) Using Mendelian randomisation to assess causality in observational studies. Evid Based Ment Health 22:67–71. https://doi.org/10.1136/ebmental-2019-300085
Bowden J, Holmes MV (2019) Meta-analysis and Mendelian randomization: A review. Res Synth Methods 10:486–496. https://doi.org/10.1002/jrsm.1346
Davey Smith G, Hemani G (2014) Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 23:R89-98. https://doi.org/10.1093/hmg/ddu328
Burgess S, Small DS, Thompson SG (2017) A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res 26:2333–2355. https://doi.org/10.1177/0962280215597579
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37:658–665. https://doi.org/10.1002/gepi.21758
Birney E (2022) Mendelian randomization. In: Cold Spring Harbor perspectives in medicine. 2022, p. 12. https://doi.org/10.1101/cshperspect.a041302
Carter AR, Sanderson E, Hammerton G et al (2021) Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol 36:465–478. https://doi.org/10.1007/s10654-021-00757-1
Davies NM, Holmes MV, Davey SG (2018) Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ (Clin Res Ed) 362:k601. https://doi.org/10.1136/bmj.k601
de Haan HG, Siegerink B, van Hylckama VA (2014) Mendelian randomisation. Ned Tijdschr Geneeskd 158:A7547
Skrivankova VW, Richmond RC, Woolf BAR et al (2021) Strengthening the reporting of observational studies in epidemiology using Mendelian randomisation (STROBE-MR): explanation and elaboration. BMJ (Clin Res Ed) 375:n2233. https://doi.org/10.1136/bmj.n2233
Wei W, Jiang W, Tian Z et al (2021) Fecal g. Streptococcus and g. Eubacterium_coprostanoligenes_group combined with sphingosine to modulate the serum dyslipidemia in high-fat diet mice. Clin Nutr (Edinburgh, Scotland). 40:4234–4245. https://doi.org/10.1016/j.clnu.2021.01.031
Yan J, Li S, Zhang Y et al (2021) Cholesterol induces pyroptosis and matrix degradation via mSREBP1-driven endoplasmic reticulum stress in intervertebral disc degeneration. Front Cell Dev Biol 9:803132. https://doi.org/10.3389/fcell.2021.803132
Wang L, He T, Liu J et al (2021) Revealing the immune infiltration landscape and identifying diagnostic biomarkers for lumbar disc herniation. Front Immunol 12:666355. https://doi.org/10.3389/fimmu.2021.666355
Han DS, Wu WK, Liu PY et al (2022) Differences in the gut microbiome and reduced fecal butyrate in elders with low skeletal muscle mass. Clin Nutr (Edinburgh, Scotland) 41:1491–1500. https://doi.org/10.1016/j.clnu.2022.05.008
Huang Y, Wang L, Zeng X et al (2022) Association of paraspinal muscle CSA and PDFF measurements with lumbar intervertebral disk degeneration in patients with chronic low back pain. Front Endocrinol 13:792819. https://doi.org/10.3389/fendo.2022.792819
Maas H, Noort W, Hodges PW et al (2018) Effects of intervertebral disc lesion and multifidus muscle resection on the structure of the lumbar intervertebral discs and paraspinal musculature of the rat. J Biomech 70:228–234. https://doi.org/10.1016/j.jbiomech.2018.01.004
Teichtahl AJ, Urquhart DM, Wang Y et al (2015) Physical inactivity is associated with narrower lumbar intervertebral discs, high fat content of paraspinal muscles and low back pain and disability. Arthritis Res Ther 17:114. https://doi.org/10.1186/s13075-015-0629-y
Feng J, Ma H, Huang Y et al (2022) Ruminococcaceae_UCG-013 promotes obesity resistance in mice. Biomedicines. https://doi.org/10.3390/biomedicines10123272
Jia J, Nie L, Liu Y (2020) Butyrate alleviates inflammatory response and NF-κB activation in human degenerated intervertebral disc tissues. Int Immunopharmacol 78:106004. https://doi.org/10.1016/j.intimp.2019.106004
Ma L, Ni Y, Wang Z et al (2020) Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut microbes 12:1–19. https://doi.org/10.1080/19490976.2020.1832857
Hu S, Wang J, Xu Y et al (2019) Anti-inflammation effects of fucosylated chondroitin sulphate from Acaudina molpadioides by altering gut microbiota in obese mice. Food Funct 10:1736–1746. https://doi.org/10.1039/c8fo02364f
Bennett DA (2010) An introduction to instrumental variables—part 2: Mendelian randomisation. Neuroepidemiology 35:307–310. https://doi.org/10.1159/000321179
Sanderson E (2021) Multivariable Mendelian randomization and mediation. In: Cold Spring Harbor perspectives in medicine, p 11. https://doi.org/10.1101/cshperspect.a038984
Smith GD, Ebrahim S (2003) “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32:1–22. https://doi.org/10.1093/ije/dyg070
Goodrich JK, Davenport ER, Beaumont M et al (2016) Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19:731–743. https://doi.org/10.1016/j.chom.2016.04.017
Goodrich JK, Waters JL, Poole AC et al (2014) Human genetics shape the gut microbiome. Cell 159:789–799. https://doi.org/10.1016/j.cell.2014.09.053
Acknowledgements
The publicly available GWAS results on gut microbia were obtained from the MiBioGen (https://mibiogen.gcc.rug.nl). The publicly available GWAS results on IVDD were obtained from the FinnGen repository (https://www.finngen.fi). We express our gratitude to the participants and research teams that permitted to build the publicly available data.
Funding
This work was supported by grants from the Natural Science Foundation of Shanxi Province (201901D111411).
Author information
Authors and Affiliations
Contributions
DZ conceived the study, carried out the data analysis, interpretation, and manuscript writing. ZW helped to draft the manuscript. LL and SC helped to check the data. JC: Methodology, Writing—review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Consent for publication
Not applicable. The data used in this study and the relevant cases were obtained from publicly available databases.
Informed consent
Not applicable. The data used in this study and the relevant cases were obtained from publicly available databases.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, D., Wu, Z., Li, L. et al. Genetic analysis of the causal relationship between gut microbiota and intervertebral disc degeneration: a two-sample Mendelian randomized study. Eur Spine J 33, 1986–1998 (2024). https://doi.org/10.1007/s00586-023-08059-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00586-023-08059-8