
Abstract
H9N2 avian influenza A viruses (AIV) have become panzootic in Eurasia over the last decade and are endemic in Iran since 1998, and inactivated vaccine has been used in chickens to control the disease. The hemagglutinin (HA), one of eight protein-coding genes, plays an important role during the early stage of infection. To study their evolution and zoonotic potential, we conducted an in silico analysis of H9N2 viruses that have infected broiler in Tehran Province, Iran between 1998 and 2007. The complete coding region of HA genes from nine H9N2 subtypes isolated from chicken flocks in Tehran Province during 1998–2007 was amplified and sequenced. Sequence analysis and phylogenetic studies of H9N2 subtype viruses on the basis of data of 9 viruses in this study and 30 selected strains are available in the GenBank. Sequence and phylogenetic analyses revealed a large number of similar substitution mutations and close evolutionary relation among sequences of HA. The isolates possessed two types of amino acid motif –R–S-S-R/G-L- and -R-S-N-R/G-L- at the cleavage site of HA. The results showed that all nine representative H9N2 isolates belong to low pathogenic AIVs since none of the amino acid sequences at the cleavage site of the HA of the isolates possessed the basic motif required for highly pathogenic viruses (R-X-R/K-R). Six out of these nine isolates possessed leucine at position 226, which prevails in the sequences found in human strains. Phylogenetic analysis showed that all our isolates belonged to the G1-like sublineage. Also, these isolates showed some degree of homology with other H9N2 isolates, e.g., 89.46–93.93.39% with qu/HK/G1/97 and 93.39–98.39% with pa/Narita/92A/98. The available evidence indicates that HA genes of H9 influenza virus circulating in Iran during the past years were not well conserved. Our finding emphasizes the importance of reinforcing AIV surveillance, especially after the emergence of high pathogenicity in poultry in Iran.
Similar content being viewed by others

Introduction
H9N2 viruses have widely circulated in the world since their first detection from turkeys in Wisconsin in1966 (Homme and Easterday 1970). H9N2 influenza viruses have become panzootic in Eurasia during the past decade and have been isolated from terrestrial poultry worldwide. The H9N2 subtype virus is a conspicuous member of the influenza family because it can infect not only chickens, ducks, and pigs but also humans (Kwon et al. 2006b). Avian influenza viruses (AIV) are segmented, negative-sense, single-stranded RNA viruses of the family Orthomyxoviridae and are divided into five genera: influenza A, B, C, Thogtovirus, and Isavirus (Fields et al. 2007). The AIV genome consists of eight segments of negative-sense, single-stranded RNA that encode at least ten proteins including two surface glycoproteins [hemagglutinin (HA) and neuraminidase (NA)], nucleoprotein, three polymerase proteins (polymerase basic 2, PB1, and polymerase acidic), two matrix (M1 and M2) proteins, and two non-structural (NS1 and NS2) proteins. Influenza A viruses are classified into a number of subtypes based on antigenic differences in their two surface glycoproteins, HA (16 subtypes) and NA (nine subtypes) (Fields et al. 2007; Harder and Werner 2006). One of the surface viral proteins, HA, undergoes frequent antigenic variation and plays a crucial role in the pathogenicity of influenza viruses. Analysis of receptor specificity of HA has indicated that HA is very important for virus transmission and is a major determinant in host range. HA is involved in the early stages of infection, causing the binding of the sialic acid receptor present on the host cell surface, and leading to fusion of the viral and endosomal membrane and subsequent entry into the host cell. The HA genes have vital relation to viral pathogenicity, antigenicity, and host range of influenza viruses, although some researchers indicated that these traits are polygenic (Jordan 2001; Swayne 2008; Webster et al. 1992).
AIVs in poultry have been categorized into two groups according to their ability to produce clinical signs and the severity of disease, the low-pathogenicity AIVs and highly pathogenic AIVs. The difference in the pathogenicity is mainly determined by the HA gene and the presence of additional basic amino acids at the proteolytic cleavage site. HA of low-pathogenicity AIVs is cleaved by trypsin-like proteases that are associated with respiratory and intestinal epithelial cells, confining the infection to these sites. In contrast, HA from HPAIVs can be cleaved by the aforementioned proteases and also by ubiquitous furin proteases, leading to a generalized infection (Alexander 2000; Jordan 2001; Swayne 2008).
Globally, there are two major, distinct gene pools of H9N2 avian influenza viruses: the North American and the Eurasian. The Eurasian lineage can be further divided into three major sublineages—the G1 lineage, represented by A/quail/Hong Kong/G1/97 (G1-like); theY280 lineage, represented by three prototype viruses A/duck/Hong Kong/Y280/97 (Y280-like), A/chicken/Beijing/1/94 (BJ94-like), and A/chicken/Hong Kong/G9/97 (G9-like); and the Korean lineage, represented by A/chicken/Korea/38349-p96323/96 (Korean-like) and A/duck/Hong Kong/Y439/97 (Y439-like) (Guan et al. 1999; Matrosovich et al. 2001); generally, the H9N2-affected chickens show mild to severe respiratory signs, edema of the head and the face, and decreased egg production accompanied with soft-shelled or misshaped eggs. The mortality is usually 5–30% depending on the type of husbandry. Coinfection of H9N2 viruses with bacteria such as Staphylococcus aureus and Haemophilus paragallinarum or with attenuated coronavirus vaccine exacerbated the disease (Jordan 2001; Xu et al. 2008).
In Iran, the H9N2 virus was first isolated from chicken in 1998 in Qazvin province and is now the most prevalent subtype of influenza virus in poultry in Iran. Mortality rates in some broiler farms in Iran are up to 65% (Nili and Asasi 2003; Nili and Asasi 2002; Vasfi Marandi et al. 2002). However, the isolate was characterized as non-highly pathogenic in the laboratory. Mixed infections of influenza virus with other respiratory pathogens have been found to be responsible for such a high mortality, resulting in great economic losses. Mosleh et al. (2009) showed that the Iranian H9N2 influenza viruses had the ability to replicate in some organs like the spleen, kidney, and other organs, which was previously unexpected for low virulence avian influenza virus. These results indicated that Iranian H9N2 virus may cause some changes in their structure (Mosleh et al. 2009). The association of high mortality in recent years and report ofH5N1 and H9N2 in wild birds in Iran raised the probability of a new genetically modified avian influenza virus (Shoushtari et al. 2008). In the present study, the HA gene of H9N2AI viruses that has been isolated from commercial chickens in Iran during 1998–2007 was sequenced. The aim of the present research was to elucidate the phylogenic and evolutionary relationships of HA genes (full length) of H9N2 among these isolates and examine the relationships among these viruses and other viruses isolated from avian in the neighboring countries or areas.
Materials and methods
Sample collection
Sample collection (tracheal swabs and fecal samples) was performed according to the standard method from suspected broiler (showed clinical signs) specimens in Tehran Province in 2007. Specimens were stored at −70°C until use. Samples were collected in a 2× phosphate buffer solution (pH 7.4) containing antibiotics (10,000 IU/ml penicillin and 1 mg/ml streptomycin sulfate) and an antifungal (20 IU/ml nystatin). Other virus isolates (four from nine) were available at the central lab of the Department of Clinical Science, Faculty of Veterinary Medicine, University of Tehran (Swayne et al. 1998; Peiris et al. 1999a).
Virus isolation
Ten-day-old specific pathogen-free (SPF) embryonated chicken eggs were inoculated. These eggs were incubated at 37°C for up to 7 days, embryonic death was monitored, and then allantoic fluid was collected under routine conditions, and the presence of viruses was determined by hemagglutination assay (HA). The identification of virus subtype was determined by a standard hemagglutination inhibition and neuraminidase inhibition tests using polyclonal chicken antisera. The allantoic fluids containing virus were harvested and stored at −70°C until use. HA test negative samples were given two more passages and tested again before being declared negative for AIV isolation. The chicken isolates produced severe disease signs such as tracheitis, respiratory congestion, and rapid mortality (Capua and Alexander 2009).
RNA extraction
Viral RNA was extracted from infected allantoic fluid using RNX reagent according to the manufacturer’s instruction. Briefly, in an RNAase-free 1.5-ml tube, 800 μl of RNX TM-Plus solution (Cinnagen, Iran) was added to 200-μl allantoic fluids. After shaking, 200 μl of chloroform was added, and the mixture was centrifuged at 14,000×g at 4°C for 15 min. Equal volume of isopropanol was added to the upper phase in a new tube. The mixture was centrifuged at 12,000×g at 4°C for 15 min. The supernatant was discarded, and 500 μl of 75% ethanol was added to the pellet. After centrifugation at 7,500×g for 10 min at 4°C, the supernatant was discarded, and the pellet was dried at room temperature for few minutes. Finally, the pellet was diluted in 20 μl DEPC water. To help in dissolving, the tube was placed in 55−60°C water bath for 10 min and stored at −70°C for RT-PCR reaction (Sambrook et al. 2001).
RT and PCR reaction
Reverse transcription was done by using oligonucleotide influenza universal primer uni12: 5-AGC AAA AGC AGG-3 with RevertAid First Strand cDNA Synthesis Kit (Fermentas, Canada). Amplification of the HA gene was carried out by PCR as described by using one pair of specific primers. Primer sequences are available upon request. The reaction mixture (50 μl) contained 5 μl of cDNA, 15 pmoles of forward and reverse primers (4 μl), and Cinnagen master mix (Cinnagen, Iran). The amplification protocol was : one step of denaturation at 94°C for 3 min, 35 cycles of 94°C/45 s, 58°C/45 s, and 72°C/60 s, and final extension at 72°C for 10 min. The PCR products were separated by electrophoresis in 1% agarose gel. PCR products were purified with the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, and USA) (Capua and Alexander 2009; Hoffmann et al. 2001; Peiris et al. 1999a).
TA cloning and sequencing
Purified PCR products for sequencing were cloned into plasmid for TA cloning with InsT/A cloning kit (Fermentas, Canada) according to the manufacturer’s instruction. Plasmid extraction from positive clone was carried out by QIA miniprep plasmid extraction kit (Qiagen, Valencia, CA, USA). Following digestion with ECORI (Fermentas, Canada) to confirm the insertion, the nucleotide sequences were analyzed by plasmid sequencing on an automated 3700 DNA sequencer (Applied Biosystems, Foster city, CA). The sequences were resolved using the ABI PRISM collection program (Perkin-Elmer, Foster City, CA) with M13 (forward and reverse) universal primer (Sambrook et al. 2001).
Phylogenetic analysis
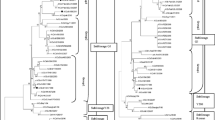
Phylogenetic analysis was carried out by analyzing the data obtained here with those of other sequences of AIV belonging to the main H9N2 lineages, (30) sequences of H9N2 viruses isolated in Iran and Asian and European countries from the GenBank database (Table 1 and Fig. 1). Nucleotide and deduced amino acid sequences were edited with the CLC Main Workbench software. Full length of the amino acid (560aa) of the HA gene was used to study and construct the respective phylogenetic trees. The phylogenetic analysis was performed with the MEGA4 (Phylogeny Inference Package) software, version 4. Distance-based neighbor-joining trees were constructed using the Tamura–Nei model available in the program MEGA, version 4. The robustness of the phylogenetic trees was assessed by 1,000 bootstrap replicates. Bootstrap values lower than 50 were omitted. The percentage similarity/difference in nucleotide sequence was estimated using CLC Main Workbench software (CLC, Denmark). All amino acids were expressed by a single-letter code. Identification and comparison o f N-glycosylation sites into reported protein sequences of HA were performed by an online server ScanProsite (http://prosite.expasy.org/scanprosite) (Dudley et al. 2007).

Phylogenetic relationships of HA genes of representative influenza A viruses isolated in Iran, Middle Eastern, Eurasian countries, and USA. The amino acid ranges of segments that were used for drawing phylogenic trees were the following: HA (1–560). Trees were generated by the neighbor-joining method with the MEGA 4 software. Numbers above branches indicate neighbor-joining bootstrap values. H9N2 viruses that were characterized in this study are indicated by red circles, Iranian pink rectangles, and Euroasian lineage green triangles. Virus name and abbreviations are shown in Table 1
Nucleotide sequence accession numbers
The amino acid sequences for all H9N2 influenza viruses used in this study are available in the GenBank under accession numbers ACA50024 through ACA50031 (presented in Table 1).
Results
A total of nine viruses were sequenced from chickens from 1998 to 2007 in Tehran Province, Iran. Nine viruses were sequenced and compared with other H9N2 sequences available in GenBank. These isolates were compared with those of some available full-length genomes of H9N2 strains isolated during the period 1966–2008 and deposited in the GenBank database. The amino acid sequences of the HA were deduced from the nucleotide sequences. The positions of each amino acid residue were numbered according to the HA sequence of human H3 subtype isolate/Aichi/2/68.
Cleavage site
The deduced amino acid sequences of the HA proteins in cleavage site were aligned and compared among the H9N2 avian influenza viruses. Amino acid sequences at the cleavage site of the HA of the isolates possessed -P-A-R-S-S-R-G-L- motif, except for two isolated: TH85 (A to T) and TH386 (S to N). All changes in region of coding cleavage site are available in Table 2.
Analysis of potential N-glycosylation
Analysis of potential N-glycosylation (glycosylation) site (PGS) motifs N-X-S/T in the HA molecule of mentioned study viruses through psoeitscan revealed seven PGSs: 1 (aa 29 to 31), 2 (aa 141 to 143), 3 (aa 218 to 220), 4 (aa 298 to 300), 5 (aa 305 to 307), 6 (aa 492 to 494), and 7 (aa 551 to 553) were all the same: NST, NVS, NRT, NTT, NVS, NGT, and NGS, respectively, except in TH79 and TH286 (21, 94, 132, 289, and 296 in HA1 and 154 and 213 in HA2 in H3 numbering; Table 3).
Receptor binding sites
In the present study, seven from nine H9N2 Iranian isolates possessed amino acid 226-L (numbered according to H3) at the receptor binding site, indicating its potential to infect humans. Of the three Eurasian-sublineage reference strains studied, amino acid 226-L is found in Qa/HK/G1/97, human isolate HK/1073/99, and the Dk/HK/Y280/97 (Wan and Perez 2007). Receptor binding sites of the isolates in this study are somewhat similar to human isolate (H3) which could be a danger. In our nine isolates, the amino acid residues of −98, 153, 155, 183, 190, 194, 195 in receptor binding sites are identical (Table 2).
Phylogenetic analysis
Evolutionary relationships of HA amino acid sequences were determined by comparing H9N2 isolates from 1998–2007 with the established Eurasian H9N2 lineages: namely, the G1, G9, Y280, and Y439 presented by their respective prototype viruses. Iranian H9N2 isolates from broiler formed a single group, which was subdivided into two subgroups.
Our study showed that the HAs of most isolates from commercial chickens in Iran from 1998 to 2007 belonged to one lineage as well as those of HK/G1/97 strains (G1-like). Also, these isolates showed some degree of homology with other H9N2 isolates, e.g., 89.46–93.93.39% with G1 and 93.39–98.39% with Narita-parakeet98.
The AA sequences of this study were compared with prototype A/turkey/Wisconsin/66. The results indicated that AA homology among these isolates with Wisconsin-66 was between 88.04% (TH78) and 88.75% (TH77). Amino acid comparisons of isolates that were sequenced in this study showed a range of minimum homology (91.1%) between TH286 and TH81 isolates to maximum homology (99.11%) between TH186 and TH286.
Discussion
In recent years, avian influenza (AI) has caused major economic harms in Iranian poultry industry. The latest Iranian H9N2 isolate has been reported to be low pathogenic for SPF chickens (data not shown). However, recent H9N2 outbreaks have caused up to 65% mortality rate and are the only influenza subtype in the chicken population in Iran (Nili and Asasi 2003; Nili and Asasi 2002).
Although the pathogenicity of avian influenza viruses is a polygenic property, the amino acid sequence of the connecting peptide of HA is considered to be its major determinant. The pathogenicity of H5 and H7 influenza viruses is associated with the presence of multiple basic amino acids at the cleavage site of HA, yet no motif standards have been established to evaluate the high or low pathogenic H9 subtype viruses. However, Guo et al. supposed that only one nucleotide substitution could change serine to arginine and produce the basic motif required for highly pathogenic viruses (Guo et al. 2001). However, the presence of this particular motif also emphasizes that these viruses have the potential to become pathogenic should they acquire any further nucleotide substitutions in the HA-connecting peptide region. The finding suggests that the H9N2 influenza viruses circulating in poultry in Iran have the potential to become highly pathogenic. All the H9 viruses tested (except TH85 and TH386) in this study possessed the motif of -P-A-R-S-S-R/G-L-, which was identical to the sequences of Gl and Y280. All recurrent H9N2 viruses possessed the single basic amino acid at the cleavage site, and they were classified as LPAI viruses. The nucleotide sequence of the HA cleavage sites of these viruses showed that only one additional nucleotide substitution (T or C to A) is needed at two positions to change serine (S) to arginine (R) and produce the basic motif required for highly pathogenic viruses (R/K-X-R-R); cleavage site motif in TH386 is similar to isolates of Israel such as AAZ14994 and AAZ14120. PARSNRG motif is specific to isolates of Israel (Peiris et al. 1999b). This motif does not exist in Iranian isolation before 2007. Amino acid in cleavage site of TH85 (PTRSSRG) is found in isolates with accession numbers AAL65235 and ABV46459 .These isolates are in Chinese isolates group (Perk et al. 2006a). During 10 years of observation, -R-S-S-R- motif predominated among the Iranian isolates (Kianizadeh et al. 2006; Toroghi and Momayez 2006; Karimi et al. 2008), and new motifs appeared among the Iranian isolates. It is the first report of emergence of new substitution of amino acid in cleavage site of HA from Iran. Also, Fereidouni et al. (2010) detected -P-A-A-D-S-R-G-L- motif in H9N2 isolate of mallard in Iran in 2007. Ghadi et al. report two new motifs, -P-A-R-S-N-K-R-L-, in isolates of live bird marketing in Iran. However, the presence of this particular motif also emphasizes that these viruses have the potential to become pathogenic should they acquire any further nucleotide substitutions in the HA-connecting peptide region. These isolates show some levels of substitution, indicating antigenic drift (Ghadi et al. 2010).
Glycosylation of HA and NA can affect the host specificity, virulence, and infectivity of an influenza strain either directly, by changing the biological properties of HA and NA, or indirectly, by attenuating receptor binding, masking antigenic regions of the protein, impeding the activation of the protein precursor HA0 via its cleavage into the disulfide-linked subunits HA1 and HA2, and regulating catalytic activity (Sun et al. 2011; Gallagher and Henneberry 1992). Generally, H9 viruses vary in the number of glycosylation sites on HA (six in duck viruses and as many as eight in quail and chicken viruses) (Xu et al. 2008). The glycosylation at position 58 was observed among H5 and H7 viruses isolated from chickens but not from aquatic birds, and it was suggested to be a feature of chicken viruses (Kwon et al. 2006a). One potential glycosylation site at position 218 (210 in H3) was lost compared with representative reference strains of the G1 lineage, the Y280 lineage, the Korean lineage, and the prototypic Pakistani isolate Ck/Pakistan/2/99. Loss of an additional glycosylation site at position 206, compared with the prototype G1 virus A/Qa/HK/G1/97, was observed; this was also seen in H9N2 viruses from the Middle East in 2000–2003. The loss of potential glycosylation sites may represent a selected adaptation of H9N2 within poultry since alteration in glycosylation pattern has been suggested to influence adaptation of avian influenza viruses to poultry (Iqbal et al. 2009). Also, deletion of one amino acid residue at position 298 in TH286 and 492 in TH79 led to the loss of one potential glycosylation site on the HA molecule . Ghadi et al. report a similar deletion in their studies on live bird marketing H9N2 isolates. They suggest that these differences represent mutations that could have occurred in markets; there was no additional glycosylation site that could induce virus virulence (Ghadi et al. 2010). High similarity in the amino acid sequences in glycosylation sites of these nine isolates provides further evidence of their similar nucleotide sequences.
A major determinant of host range is the affinity of the viral HA protein to the sialic acid (SA) receptor of the host cell. A change of preference for the avian influenza virus for a (2, 3)-linked Gal to a (2, 6)-linked Gal for the SA receptor is highly related to host specificity. Wan and Perez (2007) also demonstrated that the Glu to Leu substitution at amino acid position 226 in HA allows H9N2 viruses to replicate more efficiently in human airway epithelial cells cultured in vitro. The substitution of Gln for Leu at residue 226 and the change of Ala to Thr at position 190 occurred in the receptor binding site of those H9N2 variants that have previously been reported to be involved in the binding specificity to receptors in host cells. Therefore, further studies are needed to understand the role of these mutations in the receptor binding site in restricting the host range of these H9N2 variants. These viruses at prevalence might have had the ability of infecting humans without pigs as intermediate hosts, which suggested that H9N2 AIV would become of greater risk to public health in the coming years (Wan and Perez 2007).
The left edge of binding pocket (aa 224–229) in this study isolates had the three types of motif, in which NGLIGR motif is similar to human isolates (Liu et al. 2004). Data have been shown in Table 2. A study about left edge of binding pocket reveals that three isolates in this study (TH186, TH286, and TH386) have motif that is similar to H9N2 isolates that were isolated from humans. This finding alerts this subject that Iranian isolates have a tendency to infect humans. This kind of motif had been seen in other Iranian isolates that were obtained from Genbank. One chicken isolate (TH85) from Tehran in 2007 contained a Q-234 instead of L-234 in HA1 receptor binding site and thus was not a potential to infect humans. L-234 substation has been seen in some sequences of HA gene of Iranian H9N2 isolates and recently in isolates TH186, TH286, and TH386; thus, these isolates can be bound to human receptors (a-2,6) and may infect human populations. This substitution has been seen in some sequences of HA gene of Iranian H9N2 isolates. Another HA marker, amino acid 198 (receptor binding site) in one isolate, TH286, conserve to new motif in Iranian isolate. Also, the right edge of binding pocket (aa134–138) in Iranian isolates has been detected .Two kinds of motif GTSKA and GTSKS were detected in this study. Viruses that were isolated after 2006 show new motif in this region (TH85, TH186, TH286, TH386) (Liu et al. 2004; Lu et al. 2005).
Signal peptide (aa8), bottom of globular head (aa127), and adjacent receptor binding site (aa164) are three important positions for characterization of HA (Lu et al. 2005). These positions are available in Table 2.The mentioned positions are A, N(R), and H, respectively, in duck isolate. According to our results, TH85 (base on bottom of globular head position) and TH78 (based on adjacent receptor binding site) may be originated from ducks. Overall, the presence of H9N2 viruses of the sequence known to bind to human-type receptors in these Iranian poultry and the presence of H9N2 antibodies in the human population of Iran showed that it is possible for circulating H9N2 avian influenza viruses in Iran to infect humans. Hence, extensive surveillance of H9N2 in this country is highly recommended. According to the phylogenetic analysis of H9 genes, the H9N2 Iranian isolates fell into a single G1-like lineage, together with all of the Middle Eastern and some Asian viruses that were examined in the present study.
The phylogenetic analysis based on the amino acid sequence of a HA region showed that the isolates from Iran, Pakistan, India, and Saudi Arabia clustered together, indicating a close relationship. Interestingly, the isolates from each particular country formed a homogenous group, revealing that these viruses probably had a single source (Banks et al. 2000; Aamir et al. 2007; Zhao et al. 2011; Perk et al. 2006b; Butt et al. 2011; Tosh et al. 2008). These results are basically in agreement with previous studies. The presence of the Middle East isolates on this lineage may be due to a close geographical relationship between these countries that had given the impression that the outbreaks seen in these countries were the result of a major epizootic caused by the spread of a single virus.
The first scientific report of phylogenetic analysis of Iranian H9N2 isolates was done by Karimi et al. Nucleotide sequence comparisons of HA gene from Iranian isolates showed 97–99% identity within the group and 98% homology with the two isolates [A/Parakeet/Narita/92A/98 (H9N2)] and [A/Parakeet/Chiba/1/97 (H9N2)] from Pakistani parakeets imported to Japan (Karimi et al. 2004) Toroghi et al. report that the amino acid sequences of the isolates in 2000–2001 were very similar to those from Saudi Arabia, Germany, and Pakistan. It is postulated that, except for some Chinese isolates, the pathogenicity of Iranian isolates seems to be similar to that of other Eurasian isolates (Toroghi and Momayez 2006). Phylogenetic analysis that was carried out by Homayounimehr showed that all their H9N2 isolates from commercial chickens in Iran (1998–2002) belonged to the G1-like sublineage. On the basis of phylogenetic and molecular characterization evidence, they concluded that the H9N2 subtype influenza viruses circulating in chicken flocks in Iran since 1998–2002 had a common origin (Homayounimehr et al. 2010). Ghadi et al. in a molecular surveillance of H9N2 in live bird market report that H9N2 viruses isolated from live bird markets were highly similar to viruses isolated from industrial poultry being circulated as early as 2001. They suggest that a common source of H9N2 viruses is circulating in Iran (Ghadi et al. 2010). Shahsavandi et al. analyzed the full-length HA genes of 287 H9N2 AI strains isolated from chickens in Asia during the period 1994–2009. They showed that G1-like viruses circulated in the Middle East and Indian subcontinent countries, whereas other sublineages existed in Far East countries. It also revealed G1-like viruses with an average 96.7% identity clustered into two subgroups largely based on their time of isolation (Shahsavandi et al. 2011). In a recent study by Feriedouni et al. on avian influenza virus monitoring in wintering water birds in Iran, 2003–2007, isolate IR-Mallard-07 [A/mallard/Iran/C364/2007(H9N2))] was located in Y-439. A/Garganey/Iran/G8/2003 (H9N2) was isolated from a Garganeyin, the southern part of Iran which was suspected to have been kept together with backyard poultry for a short time before sampling (Fereidouni et al. 2010).
Our present study has demonstrated that low-pathogenic H9N2 influenza viruses have been permanently circulating in the Middle East region, including Iran, during the last decade. According to a previous report, on the basis of phylogenetic evidence, it is proposed that the emergence of H9N2 avian influenza infection in Iran originated in Pakistan, and it was due to low quarantine measures in the international boundaries. Due to the high percentage of H9N2 homology isolates of Iran with other isolates, namely A/quail/HongKong/G1, in GenBank and based on published reports for high similarity with infecting human H5N1 isolates, it seems that the potential of Iranian avian influenza isolates to infect human should be considered. Live bird markets as already mentioned are believed to be one of the best environments for keeping avian influenza viruses and transmission between species and productive source for mixing and reassortment of new viruses with higher pathogenicity. It is possible that an elevation in mortality rate under field condition could be caused by coinfection of recent isolates with the bacteria such as Mycoplasma, Escherichia coli, and Ornithobacterium rhinotracheale rather than by an emerging pathogenic H9N2 subtype of the virus. The present findings also indicate that the HA genes of the H9 influenza virus circulating in Tehran province were not also well conserved and in recent years have dominant changes. Especially from 2003, a dominant change had been produced in Iranian isolates. Continuous surveillance would improve our understanding of the role of various avian hosts in ecology of influenza viruses and thus the underlying phenomena in emergence of pandemic strains. Also, according to an H5N1 case report in non-industry birds, it is very critical for H9N2 monitoring for probable changes in H9N2 isolates.
References
Aamir UB, Wernery U, Ilyushina N, Webster RG (2007) Characterization of avian H9N2 influenza viruses from United Arab Emirates 2000 to 2003. Virology 361:45–55. doi:10.1016/j.virol.2006.10.037
Alexander DJ (2000) A review of avian influenza in different bird species. Vet Microbiol 74:3–13
Banks J, Speidel E, McCauley J, Alexander D (2000) Phylogenetic analysis of H7 haemagglutinin subtype influenza A viruses. Arch Virol 145:1047–1058
Butt AM, Siddique S, Tahir S et al (2011) Comparative sequence, antigenic and phylogenetic analysis of avian influenza (H9N2) surface proteins isolated in Pakistan between 1999 and 2008. J Infect Dev Ctries 5:413–424
Capua I, Alexander DJ (2009) Avian influenza and Newcastle disease: a field and laboratory manual. Springer, Milan
Dudley K, Nei M, Kumar S (2007) MEGA4: Molecular evolutionary genetics analysis software—version 4.0. Mol Biol Evol 24:1596–1599
Fereidouni SR, Werner O, Starick E et al (2010) Avian influenza virus monitoring in wintering waterbirds in Iran, 2003–2007. Virol J 7:1–14
Fields BN, Knipe DM, Howley PM (2007) Fields virology, vols 1 and 2. Lippincott, Philadelphia
Gallagher P, Henneberry J (1992) Glycosylation requirements for intracellular transport and function of the hemagglutinin of influenza virus. J Virol 66(12):7136–7145
Ghadi S, Bozorgmehri-Fard M, Karimi V et al (2010) Genetic analysis of hemagglutinin protein of H9N2 isolated from live bird markets in Tehran Province in 2007–2008. IJV 3:7–15
Guan Y, Shortridge KF, Krauss S, Webster RG (1999) Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proceedings of the National Academy of Sciences of the United States of America 96:9363–9367
Guo Y, Dong J, Wang M et al (2001) Characterization of hemagglutinin gene of influenza A virus subtype H9N2. Chin Med J 114:76–79
Harder TC, Werner O (2006) Avian influenza. Influenza report 48–87. Flying Publisher, Paris, Cagliari, Wuppertal, Sevilla
Hoffmann E, Stech J, Guan Y et al (2001) Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289
Homayounimehr AR, Dadras H, Shoushtari A, Pourbakhsh SA (2010) Sequence and phylogenetic analysis of the haemagglutinin genes of H9N2 avian influenza viruses isolated from commercial chickens in Iran. Trop Anim Health Prod 42:1291–1297
Homme P, Easterday B (1970) Avian influenza virus infections. I. Characteristics of influenza A/Turkey/Wisconsin/1966 virus. Avian Dis 14(1):66–74
Iqbal M, Yaqub T, Reddy K, McCauley JW (2009) Novel genotypes of H9N2 influenza A viruses isolated from poultry in Pakistan containing NS genes similar to highly pathogenic H7N3 and H5N1 viruses. PLoS One 4:e5788
Jordan F (2001) Poultry diseases. Saunders, London
Karimi V, Ghalyanchi Langeroudi A, Fard MHB, Mahboudi F, Barin A, Kheiri MT (2008) Amino acid sequence analysis of hemagglutinin protein of H9N2 isolated from broilers in Tehran in 2007. IJV 1:15–19
Karimi V, Fard MHB, Shahbazzadeh D et al (2004) Sequence analysis and phylogenetic study of hemagglutinin Gene of H9N2 subtype of avian influenza virus isolated during 1998–2002 in Iran. Iran Biomed J 8:167–172
Kianizadeh M, Pourbakhch SA, Toroghi R, Momayez R (2006) Pathogencitiy and haemagglutinin gene sequence analysis of Iranian avian influenza H9N2 viruses isolated during 1998–2001. IJVR 7:37–41
Kwon H-J, Cho S-H, Kim M-C et al (2006a) Molecular epizootiology of recurrent low pathogenic avian influenza by H9N2 subtype virus in Korea. Avian Pathol 35:309–315. doi:10.1080/03079450600821166
Kwon HJ, Cho SH, Kim MC et al (2006b) Molecular epizootiology of recurrent low pathogenic avian influenza by H9N2 subtype virus in Korea. Avian Pathol 35:309–315
Liu J-H, Okazaki K, Mweene A et al (2004) Genetic conservation of hemagglutinin gene of H9 influenza virus in chicken population in Mainland China. Virus Genes 29:329–334. doi:10.1007/s11262-004-7436-x
Lu J-H, Liu X-F, Shao W-X et al (2005) Phylogenetic analysis of eight genes of H9N2 subtype influenza virus: a mainland China strain possessing early isolates’ genes that have been circulating. Virus Genes 31:163–169. doi:10.1007/s11262-005-1790-1
Matrosovich MN, Krauss S, Webster RG (2001) H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Virology 281:156–162
Mosleh N, Dadras H, Mohammadi A (2009) Evaluation of H9N2 avian influenza virus dissemination in various organs of experimentally infected broiler chickens using RT-PCR. Iran J Vet Res 10:12–20
Nili H, Asasi K (2003) Avian influenza (H9N2) outbreak in Iran. Avian Dis 47:828–831
Nili H, Asasi K (2002) Natural cases and an experimental study of H9N2 avian influenza in commercial broiler chickens of Iran. Avian Pathol 31:247–252
Peiris M, Yam W, Chan K et al (1999a) Influenza A H9N2: aspects of laboratory diagnosis. J Clin Microbiol 37:3426
Peiris M, Yuen K, Leung C et al (1999b) Human infection with influenza H9N2. Lancet 354:916–917
Perk S, Banet-Noach C, Shihmanter E et al (2006a) Genetic characterization of the H9N2 influenza viruses circulated in the poultry population in Israel. Comp Immunol Microbiol Infect Dis 29:207–223
Perk S, Panshin A, Shihmanter E et al (2006b) Ecology and molecular epidemiology of H9N2 avian influenza viruses isolated in Israel during 2000–2004 epizootic. Dev Biol 124:201–209
Sambrook J, Fritsch EF, Maniatis T et al (2001) Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Shahsavandi S, Salmanian A-H, Ghorashi SA et al (2011) Evolutionary characterization of hemagglutinin gene of H9N2 influenza viruses isolated from Asia. Res Vet Sci. doi:10.1016/j.rvsc.2011.07.033
Shoushtari A, Hablolvarid M, Vascellari M, Hedayati A (2008) Mortality of wild swans associated with naturally infection with highly pathogenic H5N1 avian influenza virus in Iran. Archives of Razi 62:207–213
Sun S, Wang Q, Zhao F (2011) Glycosylation site alteration in the evolution of influenza A (H1N1) viruses. PloS One 6(7):e22844
Swayne DE (2008) Avian influenza. Wiley, Hoboken
Swayne DE, Glisson JR, Jackwood MW et al (1998) A laboratory manual for the isolation and identification of avian pathogens. American Association of Avian Pathologists, Philadelphia, PA
Toroghi R, Momayez R (2006) Biological and molecular characterization of avian influenza virus (H9N2) isolates from Iran. Acta Virol 50:163–168
Tosh C, Nagarajan S, Behera P et al (2008) Genetic analysis of H9N2 avian influenza viruses isolated from India. Arch Virol 153:1433–1439. doi:10.1007/s00705-008-0131-9
Vasfi Marandi M, Bozorg Mehrifard MH, Tabatabaei SM (2002) A seroe-pidemiologic study of avian influenza (H9N2) in Iran. SRIV 8:23–34
Wan H, Perez DR (2007) Amino acid 226 in the hemagglutinin of H9N2 influenza viruses determines cell tropism and replication in human airway epithelial cells. J Virol 81:5181
Webster RG, Bean WJ, Gorman OT et al (1992) Evolution and ecology of influenza A viruses. Microbiol Mol Biol Rev 56:152
Xu X-J, Xu G-Y, Zhou H-B et al (2008) Evolutionary characterization of influenza virus A/duck/Hubei/W1/2004 (H9N2) isolated from central China. Virus Genes 36:79–83. doi:10.1007/s11262-007-0123-y
Zhao J, Chai L, Wang Z (2011) Sequence and phylogenetic analysis of the haemagglutinin genes of H9N2 avian influenza viruses isolated in central China during 1998–2008. Bing Du Xue Bao 27:122–128
Acknowledgment
This project was financially supported by the Research Council of Tehran University (Iran) (grant no. 75018013/6/3). The authors gratefully acknowledge the extensive support of The Biotechnology Research Center, Pasteur Institute of Iran and especially Dr. Fereidoun Mahboudi. We thank Dr. M. Razazian for providing AI suspected broiler specimens and Dr. Sasan Fereidouni for help in primer designing.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ghalyanchi Langeroudi, A., Karimi, V., Tavasoti Kheiri, M. et al. Full-length characterization and phylogenetic analysis of hemagglutinin gene of H9N2 virus isolated from broilers in Iran during 1998–2007. Comp Clin Pathol 22, 321–330 (2013). https://doi.org/10.1007/s00580-012-1405-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00580-012-1405-x