
Abstract
This study explored the tribological phenomena under minimal load using molecular dynamics. In the simulation, <NVT> ensemble average standards and the Condensed-phase Optimized Molecular Potential for Atomistic Simulation Studies potential energy function were used. Regarding materials, this study used (5, 5) carbon nanotubes (CNTs) to move the copper atoms in order to obtain the coefficient of friction (COE) between the single-wall CNTs and copper atoms before analyzing the tribological phenomena under minimal load and changes in friction. This study found that, under an extremely tiny load, the direction of the resistance of the relative sliding is not always in the opposite direction of the movement direction. The influence of environment temperatures on the COE was that the environment temperature increases, the number of fluctuation also increases, but the COE reduces.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The development of nanotechnology has included biomedical, physical, chemical, motor, and optoelectronic areas. Nanotechnology is different from traditional processing processes, as it is a process from small to large, and starts with the accumulation of atoms. From the basic units of nano particles, nano tubes, and nano films, the emerging nanotechnologies are thus developed. In addition to being a popular field of study, nanotechnology is also one of the important technologies supported by both government and industries in recent years. The development of nanotechnology has great potentials in both academia and practical application.
Carbon nanotube (CNT) is a material of nanometer scale, which has a complete molecular structure. It is viewed as the hollow seamless tubular structure consisting of curled graphite sheet formed by carbon atoms and has the geometry of a high aspect ratio and consistent radius. In addition, CNT is composed of the hexagonal carbon rings. However, in the curling and the hemisphere parts of the tube end, it contains some pentagonal and heptagonal carbocyclic ring structures. Due to its good structural properties and carbon-atom bonding by stable covalent bonding force, CNTs have mechanical properties of excellent tensile, flexural, high elasticity, and high toughness. Due to the characteristics of high aspect ratio, small girth curvature, and high durability of CNTs (Nguyen et al. 2005; Ajayan et al. 1994), the resolution and usage times of the images obtained during nano print detection experiments can be greatly enhanced. Hence, when used as a probe, CNTs have a distinct advantage. On the other hand, CNTs could replace conventional probes, as they can bend to avoid damaging the scanned specimen by forward or lateral impact.
Dai et al. (1996) applied CNTs in the atomic force microscope to significantly enhance the resolution and service life of the atomic force microscope. This manual method has been used to combine CNTs and silicon probes. However, the combination process is very time consuming; thus, the method of directly using CNTs in a probe has been developed. Although a large number of CNTs can be produced on the probe, how to effectively control the size, quantity, and growth direction of the CNTs produced using the synthesis method remains a problem. The methods of combining CNTs and probes, as well as recent relevant studies, are briefly summarized and introduced as follows.
Studies on molecular dynamics can be traced back to the statistical mechanics of Alder and Wainwright (1957), while studies on the application of friction are developed in recent years. Harrison et al. (1992) reported the atomic-scale friction phenomenon of diamond surfaces using Molecular-dynamics simulations. The friction which occurs when two diamond (111) hydrogen-terminated surfaces are placed in sliding contact is investigated for sliding in different crystallographic directions, as a function of applied load, temperature, and sliding velocity. But they did not investigate the friction or the tribological properties between copper and carbon material (such as diamonds or CNT). Pokropivny et al. (1997) used a tungsten carbide probe to slide over the surface of BCC-iron in a study of the adsorption wear mechanism at the atomic level of the friction process. Adsorption wear could be attributed to lack of attractiveness of probe atoms and the substrate, resulting in the attraction of atoms at the probe tip and substrate surface, which may cause wear and tear. Shimizu et al. (1998) used a hard diamond as the probe and single crystal copper as the substrate in a friction experiment at the atomic level. The stick–slip phenomenon at the atomic level was observed, which proved that using simulation of molecular dynamics could observe the phenomena observed by the atomic force microscope. They also discussed probe cantilever rigidity and stick–slip phenomena. Mikulla et al. (1998) used molecular dynamics for simulation of the sliding behavior of two 2D copper tablets, and found the generation of the dislocation loops of different levels under the sliding surface. Sorensen et al. (1996) used copper atoms as the material to study the mechanical properties between the probe, the surface, and between the surfaces. The results showed that friction was generated by the stick–slip phenomena at the atomic level, which was caused by the nucleation of atoms and the dislocation phenomena.
In recent years, CNTs have been a very popular research direction, which are formed by sp2 in the carbon atoms through bonding. Its special dimensions and structure have granted CNTs with unique material characteristics and wide applications. CNTs can be used as the carbon probe for AFM. In general, the pressure is lower under AFM; however, friction may affect the data. Therefore, this study explored the tribological phenomena of CNTs under minimal load.
2 Physical model and MD simulation related parameters
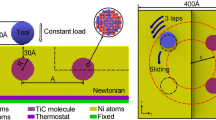
In this study, the work piece was single crystal copper and the slider was presumably formed by CNTs as shown in Fig. 1. The diameter of the CNT is 6.758 nm. In MD simulation, the work piece was divided into a freely movable area and a fixed boundary layer (Chang et al. 1993). The simulation space was a copper work piece. The selection of copper material was due to the importance of copper in the nanometer process, and application of copper as the interconnect metal in semiconductor technology. In Fig. 1, for simulation of the nanometer tribology on the surface of the copper work piece (001), the two layers of atoms on the bottom XY plane of the work piece were fixed, while the other atoms of the work piece were allowed to move in MD simulation.

Simulation of nanometer model using molecular dynamics
In order to simplify the simulation situation to faster simulation speed, the simulation environment is set a vacuum environment in this study. Therefore, the CNT model was constructed for the clean-cut CNT rather than a capped end or a hydrogen-saturated end in contact with the copper.
The environment temperature would seem to be an important parameter for tribological results, therefore, the environment temperature is at 300, 350 and 400 K in order to investigate the temperature effect in this study.
The time step significantly affects the reliability of the results of molecular dynamics simulation, and too small a time step requires an enormous amount of calculation, while a larger time step leads to unreliable results; 1 femtosecond (fs, 1 × 10−15 s) is only about 1/300 of the vibration cycle of the copper atoms (the atomic vibration period of copper at room temperature is about 3 × 10−13 s (Ma and Yang 2003). Therefore, the simulation time step is 1 fs, which can provide sufficient accuracy.
3 Simulation process
The simulation in this study was conducted according to the <NVT> ensemble average standards and the time step was 1 fs. By the continuous calculation of different time periods, a new speed, force, and location of each copper and carbon atom until the set dynamic simulation time were obtained. After the establishing the molecular dynamic model of the CNT, simulation was conducted on the arrangement of copper and carbon atoms upon reaching the lowest possible energy position, in order to avoid the divergence caused by instable system energy in late Molecular dynamic simulation (Haile 1992).
In the simulation, the initial speed of each carbon atom of the system was determined by the Boltzmann probability distribution (Haile 1992). Afterwards, the potential energy function differential was determined to obtain the interaction forces between atoms. According to Newton’s second law, the system acceleration of the next time step was determined. The speed and position of each atom in the system was determined by the Velocity-Verlet integral algorithm iterative solution (Swope William et al. 1982). In this simulation, for each displacement of CNT (X-axis or Z-axis), 200 times the MD balance calculation were conducted at the original position to obtain the actual data of the position.
4 Selection of potential energy function and related parameters
This study used Condensed-phase Optimized Molecular Potential for Atomistic Simulation Studies (COMPASS) as the potential energy function for CNT simulation. COMPASS (Sun 1998) is the force field in support of the atomic level computation of solid or liquid materials. It is the ab initio force field of the parameterization and verification of the first solid or liquid properties, and the ab initio principles of molecules and experimental data. It can accurately predict the molecular system, the molecular structure, and types of liquid and solid systems, vibration, and thermo-physics properties in the temperature and pressure range of simulation. The mathematical model of the potential energy function is as shown in Eq. (1). The constants of the COMPASS potential energy function in benzene-like ring fullerene structures (c3a) of the single-wall CNTs are as shown in Table 1.
The above equation includes the bonding, non-bonding, and the interactive coupling items, as follows:
ϕ (r) COMPASS for the simulation of the COMPASS potential energy function, b 0 ideal bond length, K 2, K 3, K 4 bond stretching force constant, θ0 ideal bond bending angle, H 2, H 3, H 4 bond bending force constant, ϕ ideal internal plane torsion angle, V 1, V 2, V 3 bond torsion force constant, χ external plane torsion angle, K x external plane torsion force constant, K bθ the interactive coupling force constant of bonds and bending, (b 0) an ideal bond length of bonds in another direction, K bb′ the interactive coupling force constant of bonds, K 1bϕ, K 2bϕ, K 3bϕ the interactive coupling force constant of bonds and torsion, K 1θϕ, K 2θϕ, K 3θϕ the interactive coupling force constant of bonds and torsion, q i, q j i and j atomic charge, e Coulomb constant, σ atomic diameter, εij relative energy unit between atoms i and j, r ij the relative distance between atoms i and j
The optimization of the atomic potential arrangement can obtain the actual physical structure, thus, saving the time to reach a convergence of temperature and energy in the simulation of system atoms. Therefore, after the establishment of the CNT model, this study used three methods of optimization (Ma and Yang 2003), including Steepest Descents Method, Conjugate Gradient Method, and Newton–Raphson Method, in order to minimize the lowest potential energy of the carbon atomic arrangement within the lattice system of the single-wall CNT. The optimization’s design variable is the location of the carbon atom arrangement, while the target function is the lowest potential energy of the carbon atom arrangement within the lattice system of the single-wall CNT, and the times of iteration is about 5,000 times.
Prior to simulation, the arrangement of carbon atoms should minimize the potential energy in order to reduce the impact between atoms at the lowest level and maintain the potential energy gradient to “0”, as possible. The structure after the optimization arrangement is more consistent with the actual physical structure.
In this paper, the Morse potentials are adopted for the calculation process at copper–copper interactions, and for Cu–C (CNT) interactions.
It is
where ϕ(r ij ) is a pair potential energy function, D is the cohesion energy, α is the elastic modulus and r 0 is the particle distance at equilibrium. The related parameters are shown in Table 2.
5 Calculating the action force and coefficient of friction
The method for calculating the action force in terms of the repulsive forces and the attractive forces caused by the potential energy between the atoms of the CNT and every atom of the workpiece are added up in the three axial directions Fix, Fiy and Fiz, to yield the three axial directions Fx, Fy and Fz.
The calculation of action force and the formulas are as follows:
Among which, i is the number given to the carbon atom on the tool, j is the number given to the copper atom in the material, m is the quantities of carbon atoms in the CNT, n is the quantities of copper atoms in the material, and r ij is the distance between Number j copper atom and Number i carbon atom in the CNT.
The calculation of coefficient of friction (COE) and the formulas are as follows:
6 Results and discussion
The carbon atoms at one end of the CNT were fixed only on the Z-axis. The CNT was dropped along the direction of the Z-axis for 40 times at the rate of 0.02 Å/fs until the atom at the tip of the CNT was at a distance 1.5 Å from the work piece surface by copper atom. In the simulation, each displacement of the CNT (X-axis or Z-axis) led to 200 times of MD balance calculation of the entire system at the original location to obtain the actual data of the location. The three axial forces in simulation are as shown in Fig. 2. As shown in Fig. 2, when the CNT gradually approached the work piece surface, the Z-axis force of the CNT increased, indicating that the copper atoms on the surface of the work piece and CNTs have generated repelling force. A special phenomenon was observed in Fig. 2. When the CNT did not move horizontally, the forces on X and Y axis were not zero. Instead, when the CNT gradually approached the work piece surface, the forces on X and Y axis increased accordingly. This is because of the hollow structure of the CNT (as shown in Fig. 3). Therefore, the relative position of the carbon atom at the tip of CNT and work piece surface copper atom on XY axis may not be likely at the location of the force balance. Therefore, when the CNT gradually approached the surface of the work piece, the force between the carbon atom at the tip of the CNT and the copper atom on the surface of the work piece (repulsion or attraction) may increase accordingly. Therefore, when the CNT gradually approached the surface of the work piece, the forces on X and Y axis would increase accordingly.

The three axial forces of the CNT decreasing 40 steps along the direction of Z-axis

Relative positions of the carbon atom of the CNT and copper atoms on work piece surface on XY axis (top view)
When the distance between the atom at the tip of the CNT and the work piece surface was 1.5 Å, the movement by 0.02 Å/fs on X axis was conducted to explore the tribological behavior of CNTs on the surface of copper work piece. The simulation results are as shown in Fig. 4, and the three axial forces by simulation are as shown in Fig. 5. As shown in Fig. 5, force on Z axis decreased and tended to be stabilized with increasing number of movement steps of the CNT on X axis. This is caused by the compression of the CNT on Z axis. Due to the horizontal displacement, the compression on the Z axis slightly relaxed before displaying a stable force. As shown by the arrows in Fig. 6, the atoms of CNTs deviated from the original axis. According to Fig. 5, the three-axial forces were changing in a certain pattern when the CNT movement along X axial direction for 500 time steps. This study suggested that, when the X axis was moving, due to the hollow structure of the CNT and the interval between the copper atoms on the work piece surface, the force between carbon atoms at the tip the CNT and the copper atoms (repulsion or attraction) may not be likely in the position of the force balance. Therefore, with the movement of the CNTs, forces on X axis and Y axis would fluctuate, and the fluctuation of the force was related to the lattice of copper atoms.

Tribological behavior of the CNT on the surface of the copper work piece a No. 250 step on X axis and b No. 500 step on X axis

The three-axial forces when the CNT moving along X axial direction for 500 time steps

The deviation from the original axis of the atoms of the CNTs
The COF was obtained by dividing the X axial force by Z axial force of each step. The result is as shown in Fig. 7. As shown in Fig. 7, under an extremely tiny load, the COF of the CNT against the copper surface changed, and may have negative values.

The coefficient of friction at each time step under CNT moving along X axial direction at 300 K
In the macroscopic scale, the direction of friction force usually associated with the movement of objects in the opposite direction, and the friction force is the resistance. Traditionally, the friction force (resistance force) or COF is set to a positive value. As the movement of CNT, the action force on the X-axis and Y-axis will be ups and downs. That is not only a phenomenon of repulsive force, will also have attractive force appears. When the attractive force occurs, which means that at this time the friction force is not resistance, instead, the force is attractive force and helps the objects forward, then the friction force is negative, it will appear a negative COE. Therefore, under an extremely tiny load, the direction of the resistance of the relative sliding is not always in the opposite direction of the movement direction, for reasons as described in the above paragraph.
The environment temperatures ranging from 300 to 400 K with a contact distance of the 1.5 Å between CNT and the work piece surface was executed to examine the influence of environment temperatures on the COE. The COE is plotted versus steps of CNT moving along X axial direction at different environment temperature in Fig. 8.

The coefficient of friction at each time step under CNT moving along X axial direction at 300, 350 and 400 K
From the Fig. 8, it can be found that the influence of environment temperatures on the COE was that the environment temperature increases, the number of fluctuation also increases, but the COE reduces.
7 Conclusions
This study used the molecular dynamics simulation software Materials Studio to explore the tribological phenomena under minimal load. The simulation used (NVT) ensemble average standards, the COMPASS potential energy function, and (5, 5) CNTs. By moving toward the copper atoms at the rate of 200 m/s, the COE between the single-wall CNT and copper atoms was determined. Then, the tribological phenomena and changes in friction under minimal load were analyzed.
Moreover, when the CNT did not move horizontally, the forces on X and Y axis were not zero. Instead, when the CNT gradually approached the work piece surface, the forces on X and Y axis increased accordingly. Regarding relative sliding, the force on Z axis decreased before becoming stabilized with an increasing number of steps. Under an extremely tiny load, the direction of the resistance of the relative sliding is not always in the opposite direction of the movement direction.
The influence of environment temperatures on the COE was that the environment temperature increases, the number of fluctuation also increases, but the COE reduces.
References
Alder BJ, Wainwright TE (1957) Phase transition for a hard sphere system. J Chem Phys 27(5):1208
Ajayan PM, Stemphan O, Colliex C, Trauth D (1994) Aligned carbon nanotube arrays formed by cutting a polymer resin-nanotube composite. Science 265:1212
Chang X, Thompson D, Raff LM (1993) Minimum-energy reaction paths for elementary reactions in low pressure diamond-film formation. J Chem Phys 97:10112
Dai H, Hafner JH, Rinzler AG, Colbert DT, Smalley RE (1996) Nanotubes as nanoprobes in scanning probe microscopy. Nature 384:147
Haile JM (1992) Molecular dynamic simulation: elementary methods. John Wiely, USA
Harrison JA, White CT, Colton RJ, Brenner DW (1992) Molecular-dynamics simulations of atomic-scale friction of diamond surface. Phys Rev B 46(15):9700–9708
Lin Z-C, Huang J-C (2008) The study of estimation method of cutting force for conical tool under nanoscale depth of cut by molecular dynamics. Nanotechnology 19:1157011–11570113
Ma XL, Yang W (2003) Molecular dynamics simulation on burst and arrest of stacking faults in nanocrystalline Cu under nanoindentation. Nanotechnology 14:1208–1215
Mikulla RP, Hammerberg JE, Lomdahl PS, Holian BL (1998) Dislocation nucleation and dynamics at sling interfaces. Mater Res Soc Symp Proc 522:385–391
Nguyen CV, Ye Q, Meyyappan M (2005) Carbon nanotube tips for scanning probe microscopy: fabrication and high aspectratio nanometrology. Meas Sci Technol 16:2138
Pokropivny VV, Skorokhod VV, Pokropivny AV (1997) Atomic mechanism of adhesive wear during friction of atomic-sharp tungsten asperity over (114) bcc-iron surface. Mater Lett 31:49–54
Shimizu Jun, Eda Hiroshi, Yoritsune Masashi, Ohmura Etsuji (1998) Molecular dynamics simulation of friction on the atomic scale. Nanotechnology 9:118–123
Sorensen MR, Jacobsen KW, Stoltze P (1996) Simulations of atomic-scalesliding friction. Phys Rev B 53(4):2101–2113
Sun H (1998) COMPASS: an ab initial force field optimized for condensed-phase applications. J Phys Chem B 102:7338
Swope William C, Andersen HC, Berens PH, Wilson KR (1982) A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: application to small water clusters. J Chem Phys 76(1):637–649
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Huang, JC., Weng, YJ. A discussion of the tribological phenomena under minimal load using molecular dynamics. Microsyst Technol 20, 349–355 (2014). https://doi.org/10.1007/s00542-013-1916-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00542-013-1916-7