
Abstract
The human gut microbiome (bacteria, fungi, viruses, and archaea) is a complex and diverse ecosystem. It plays an important role in human health, but is involved in several intestinal and extraintestinal diseases. Most research to date has focused on the role of bacteria, while studies focusing on fungi (also referred to as “mycobiome” or “fungome”) are still in its infancy. In this review, we focus on the existing literature available about the gut mycobiome with an emphasis on compositional mycobiome changes associated with liver diseases, the impact on pathogenesis of disease, and its potential use as therapeutic targets. We also provide insights into current methodologies of studying mycobiome, and we highlight the interkingdom interactions in the context of disease and how they affect health of the host. Herein, by focusing on the gut mycobiome, this review provides novel insights and directions for liver research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human gastrointestinal tract harbors trillions of microbes such as bacteria, fungi, viruses, and archaea [1, 2]. The gut microbiome may contribute to host metabolism, health, and disease [3]. Over the past decades, extensive studies elucidated the role of bacteria in health and disease due to the higher abundance and the existence of more established reference databases [4]. In contrast, the fungal constituents of the microbiome, termed the mycobiome, are receiving less attention yet highly important in maintaining intestinal homeostasis and immunity [5]. Although fungi comprise less than 0.1% of microbial genes [6], fungal cells are 100 times larger in volume than many bacteria, thus representing a much larger biomass [7], and they may play key roles in maintaining microbial community structure and function [8]. Fungi are an ancient community group, and the fossil evidence suggests that they may be 460 million years old with the earliest fungal from Ordovician [9]. As for now, at least 99,000 species of fungi have been defined, and approximately 1,200 new species are being described every year [10]. Most fungi grow as hyphae, which are tubule-like structures measuring 2–10 μm in diameter and several centimeters in length. Fungal nuclei are found small in diameter with 6–12 chromosomes encoding for 6,000–18,000 genes [11]. Compared with bacteria, the healthy human gut mycobiome is lower in diversity and density, and it is dominated by Saccharomyces, Malassezia, and Candida [12, 13]. Studies of the human mycobiome have been focused on several body sites including skin [14], gut [15], lung [16], and oral cavity [17]. Sequencing of internal transcribed spacer (ITS) 1 region from oral cavity of 20 healthy individuals identified 74 culturable and 11 non-culturable genera, with genera including Candida, Cladosporium, Aureobasidium, an unidentified Saccharomycetales, Aspergillus, Fusarium, and Cryptococcus being the most frequent ones [17]. A second study using ITS sequencing described oral fungal composition in healthy subjects and patients with periodontal disease. Overall, at least 5 phyla were detected (Ascomycota, Basidiomycota, Glomeromycota, Chytridiomycota, and unclassified), and most of sequences were assigned to the phylum Ascomycota. Similarly, Candida and Aspergillus were the most abundant genera (detected in 100% participants). At species level, three species were observed in all participants including uncultured Dikarya, Candida spp., and Aspergillus niger [18]. Human lungs continually exchange 5–8 L/min of air that contain 102–104 fungal spores per cubic meter [19]. In healthy subjects, there was low fungal amplification, and the few fungal reads present in healthy lung samples comprised largely environmental agents such as Davidiellaceae, Cladosporium, and low abundances of Aspergillus [20]. In contrast, the human gastrointestinal tract harbors a variety of fungal genera. Sequencing of ITS1 region from 96 human fecal samples detected 66 genera. The most common genera were Saccharomyces, Candida, and Cladosporium [21]. The skin mycobiome in adults is dominated by Malassezia [22], and fungi are more diverse including organisms such as Aspergillus, Epicoccum, and Phoma in addition to Malassezia [23].
Like bacteria, fungi can be beneficial to host immunity, but they can also have deleterious effects under disease conditions. Among all fungal infections, superficial infections of the skin and nails are the most common diseases with ~ 1.7 billion individuals being affected worldwide [24]. Invasive fungal infections are less frequent yet are more deadly, affecting ~ 1.5 million people every year. More than 90% of fungal-associated mortality is caused by species from one of these four genera: Candida, Cryptococcus, Aspergillus, and Pneumocystis [25]. The gut mycobiome has recently been recognized as a novel and important player in the pathophysiology of intestinal and extraintestinal diseases [2]. It is known to have a profound influence in modulating local as well as peripheral immune responses [26]. Recent data highlight that some commensal fungal species, especially Candida albicans (C. albicans) and Malassezia spp., are potent inducers of antigen-specific T-helper cell responses in humans that are altered in inflammatory diseases, such as inflammatory bowel diseases (IBD) [27] and skin disorders [28], which thus may contribute to disease pathogenesis. The gut and liver have a bidirectional communication via the biliary system and portal vein [29]. Therefore, the gut microbiota and microbial products play critical roles in modulating liver functions. In this review, we summarize the current evidence that supports the association between gut mycobiome and various liver diseases (Table 1), such as alcohol-associated liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD). We discuss recent advances in gut mycobiome research, current methodologies and challenges to study mycobiome, and the bacteria–fungi interactions in the context of disease.
The role of fungi in host health and immunological responses
Our microbiome is a complex ecosystem, which influences diverse host functions such as metabolism, vitamin synthesis, gut permeability and function, and immunity [30]. However, understanding the dynamics and how it is controlled are challenging. Foster et al. argued that hosts are under selective pressure to control their microbiota towards the retention of beneficial species [31]. Therefore, it is possible that mycobiota is also under the selection by the host in favor of its health [4]. A number of studies revealed that the gut mycobiome plays an important role in maintaining intestinal homeostasis and systemic immunity [32], while fungal dysbiosis is associated with local and distal inflammatory diseases [26, 33]. Fungi interact with the immune system via pathogen-associated molecular pattern (PAMPs) that are recognized by innate immune receptors and are often shared between different fungal species. Recent studies show that certain fungal species can provoke cellular adaptive immune responses represented by CD4+ T lymphocytes [34]. The type of T-cell response required to control different fungal species differs remarkably [35], which becomes evident in patients with different primary immunodeficiencies [36]. Especially, Th17 cells, a subset of T-helper cells characterized by producing interleukin (IL)-17A, IL-17F, IL-21, and IL-22, have been described to be of particular importance for protecting against fungi infection. IL-17 functions through activating the IL-17 receptor, which further induces other inflammatory cytokines, chemokines, and antimicrobial peptides to exert anti-fungal activity [37]. Interestingly, patients with an impairment in Th17 immunity mainly suffer from mucocutaneous candidiasis [38], while broader fungal susceptibilities are described in patients with innate immune defects [39]. Consistently, germ-free mice colonized with C. albicans developed robust Th17 response [40], and C. albicans is a major target of human Th17 cells that can be identified in virtually all individuals [27, 41]. Induction of C. albicans-specific Th17 responses in healthy humans seems to occur during homeostatic interaction with the host in the absence of obvious inflammation. This is in line with intestinal commensal C. albicans colonization in mice, where Th17 cells are induced in the gut and mesenteric lymph nodes in the absence of any fungal infection or excessive inflammation [40, 42]. C. albicans-specific T-cell responses also broadly modulate human anti-fungal Th17 immunity via propagating Th17 cells cross-reactivity to other fungal species, such as Aspergillus fumigatus (A. fumigatus) [27], that rather generates a tolerogenic regulatory T-cell (Treg) response in healthy humans [35, 43]. These cross-reactive Th17 cells expand in patients with asthma, chronic obstructive pulmonary disease (COPD), or cystic fibrosis, contributing to fungus-induced pathological inflammation in the lung [27]. Whether C. albicans-driven cross-reactive Th17 cells also contribute to other gut-distal diseases merits further investigation. Besides C. albicans, human Th17 responses have also been described to target other fungi, especially Malassezia spp.[27, 28]. Saccharomyces cerevisiae (S. cerevisiae) spores have been described to favor a Th17 inducing environment under in vitro conditions [44]. However, overall, only few microbial species and in particular C. albicans have been identified as potent inducers of Th17 responses in humans [27], suggesting a specialized interaction pattern with the human host.
In contrast to the homeostatic interaction, Candida overgrowth is linked to several diseases, including IBD [45,46,47], alcohol-associated liver disease (ALD) [48, 49], obesity [50], and liver cirrhosis [51], and also exacerbates experimental colitis in mice [47, 52, 53]. Malassezia restricta has also been linked to IBD. In a mouse model of colitis, it exacerbates disease severity by stimulating inflammatory responses via CARD9, a signaling adaptor for anti-fungal defense as well as a genetic risk factor linked to Crohn’s disease and ulcerative colitis [47, 54]. Intestinal inflammation and epithelial damage in IBD may enhance interaction of fungi and immune cells, since C. albicans-specific Th17 responses are increased in the blood of Crohn’s disease patients [27]. Anti- S. cerevisiae antibodies (ASCA), that are cross-reactive to C. albicans, can be detected in a subset of Crohn´s patients [55] and are also elevated in patients with ALD and associated with increased mortality in patients with alcoholic hepatitis [48].
Whether fungus-specific T-cell responses are altered in ALD is currently unknown. However, increased levels of Th17 cells and their cytokines are found in blood and livers of patients with chronic liver diseases [56,57,58], and recombinant IL-17A administration induces non-alcoholic steatohepatitis (NASH) in mice [57]. In ALD, elevated serum Th17 cytokines correlated with the progression of liver damage [58], while Th17 inhibition ameliorated experimental ethanol-induced steatohepatitis [59]. Future studies are needed to investigate a potential direct interaction between C. albicans and Th17-mediated pathology in chronic liver diseases.
Similar to bacteria, fungi can also affect host metabolism and health. For example, S. cerevisiae exacerbate colitis in a mouse model by enhancing host purine metabolism and increasing the systemic level of uric acid [60]. Specific Malassezia spp. can produce metabolites acting as virulence factors that promote inflammation and further contribute to diseases, while other metabolic products may downregulate inflammatory mediator production [61]. Malassezia furfur converts tryptophan into several indole compounds, including malassezin, indirubin, and indolo [3,2-b] carbazole (ICZ), as potent ligands for the aryl hydrocarbon receptor (AhR). AhR is a nuclear receptor expressed in all skin cell types, activation of which has been linked to skin homeostasis and certain skin diseases [61]. In healthy skin, AhR signaling contributes to keratinocyte differentiation, skin pigmentation, and skin barrier function. Several studies have shown that blocking AhR signaling prevents or treats skin cancer, whereas activating AhR could be beneficial in inflammatory skin diseases [62]. Therefore, AhR signaling could be a promising target for skin diseases. Taken together, these studies show the importance of fungi and their metabolites for host health and disease. Understanding their interactions will facilitate a better understanding of disease pathogenesis and identification of targets for new therapy.
Methodologies to study the mycobiome
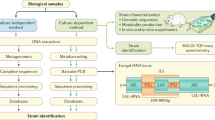
Historically, scientists have relied on culture-based methods to study the mycobiome by plating onto selective media supplemented with antibiotics to prevent bacterial growth [63]. Sabouraud dextrose media (SABDEX) supplemented with antibiotics (e.g., chloramphenicol, tetracycline, or gentamicin) is a commonly used media for culturing yeast, molds, and aciduric bacteria [2, 64, 65]. Potato dextrose media are also widely used for culturing molds and yeast [64, 66]. For culturing fast-growing fungal species, such as C. albicans, Yeast Extract–Peptone–Dextrose (YPD) media supplemented with antibiotics has been used [67]. Chromogenic media, CHROMagar™ Candida, have been developed for visual differentiation of C. albicans, C. tropicalis, and C. krusei [68]. Leeming and Notman agar can be used to culture Malassezia spp., which can be observed after incubation for 4 days [69]. Similar to what has been encountered for culturing bacteria and archaea, challenges exist for most fungal microorganisms [38]. For example, culture-based methods have a number of issues and biases, such as time-consuming, and they are biased towards fast-growing and non-fastidious species [2]. Previous studies reported that culture-based methods identified less than 30% of the fungal species from human gut compared to culture-independent methods, such as ITS1 and ITS2 sequencing [70].
To circumvent these challenges, a culture-independent method to investigate fungal communities is performing polymerase chain reaction (PCR) on extracted DNA using different primers [71]. Different regions of the fungal ribosomal DNA operon including 18S, 5.8S, 26S, and the ITS are targeted by different primers [72]. 18S region is the most widely used to study mycobiome and other eukaryotes, while it provides lower taxonomic resolution than ITS regions [73], and it might amplify non-fungal species from the host [64]. As the rapid development of next-generation sequencing, ITS regions located between 18S, 5.8S, and 28S, ITS1 and ITS2, are widely used for taxonomic identification of fungal species [73]. Despite the advantages of NGS on ITS sequencing, studies indicate that there are biases toward fungal species in terms of amplifying ITS1 or ITS2 regions [12, 32, 73]. Regardless of the methods used, high-quality reference databases are required to accurately assign fungal taxonomy [74]. Many fungal sequences submitted to GenBank have been annotated with incorrect species names, and more than 10% ITS sequences are annotated incorrectly at the species level [75]. There are three commonly used public fungal databases: the UNITE database [76], Findley database [77], and the RTL database [78]. As the choice of database will clearly affect the outcome of the study, it should be chosen with caution and tailed to the study.
Mycobiome and alcohol-associated liver disease
Alcohol-associated liver disease (ALD) represents a spectrum of liver disease ranging from simple steatosis to the more advanced alcoholic hepatitis and cirrhosis resulting from alcohol use [79]. In the United States, ALD associated mortality was estimated 5.5 per 100,000 in 2012, and the contribution to cirrhosis is trending to increase compared with hepatitis C virus [80]. Early discoveries in animal models and patients associated with ALD identified increased level of bacterial endotoxin, disrupted intestinal barrier, and increased intestinal permeability and bacterial translocation [81, 82], suggesting a link between gut microbiome and the pathogenesis of ALD. Recent studies showed that alcohol consumption is associated with decreased bacterial diversity and altered gut microbiome in animal models and human patients [83, 84]. Furthermore, intestinal virome characterization showed an increased viral diversity in fecal samples from patients with ALD compared with nonalcoholic controls, and altered viral taxa are associated with disease severity and mortality in alcoholic hepatitis patients [85]. Research in the field of mycobiome showed changes in the composition of fecal mycobiome in patients with ALD compared with non-alcoholic controls. Patients with ALD exhibited lower fungal diversity and richness and a significant overgrowth of Candida (genus) independent of stages of ALD (nonprogressive ALD, alcoholic hepatitis, or alcoholic cirrhosis). In addition, there was an increased level of serum anti-Saccharomyces cerevisiae IgG antibodies (ASCA) in alcohol use disorder (AUD) patients with simple steatosis or progressive ALD [86] as well as in patients with alcohol-related cirrhosis compared with controls, indicating an increased systemic exposure to intestinal fungi or their products in response to alcohol. More importantly, serum ASCA level is correlated with mortality in patients with alcohol-associated cirrhosis or alcoholic hepatitis [48, 49]. Increased fungal overgrowth and plasma 1,3-β-D-glucan (cell wall polysaccharides from most fungi) were observed in mice following 8 weeks of chronic ethanol administration, compared with control diet-fed mice. Treating mice with anti-fungal agent amphotericin B reduced fungal overgrowth and 1,3-β-D-glucan translocation, and attenuated ethanol-induced liver disease via reducing C-type lectin-like receptor CLEC7A dependent IL-1β production [49]. Candida albicans (C. albicans) is a commensal fungus in the human gut, our previous study suggested that its relative proportion was increased in patients with AUD and alcoholic hepatitis patients. The proportion of patients with alcoholic hepatitis positive for fecal candidalysin, an exotoxin produced by C. albicans, is higher as compared with non-alcoholic controls or patients with alcohol use disorder. Candidalysin exacerbates ethanol-induced liver disease in mice by causing direct hepatocyte damage. Alcoholic hepatitis patients that are positive for candidalysin coding gene ECE1 (extent of cell elongation 1) exhibit more severe disease and higher mortality [67]. Notably, candidalysin does not increase intestinal permeability or cause intestinal epithelial cell damage in ethanol-fed mice. It is possible that candidalysin reaches the liver from intestinal lumen via increased intestinal permeability [67]. Candidalysin can directly damage hepatocytes in culture.
The genus Penicillium dominated the mycobiome of nonalcoholic controls, and the abundance was negatively correlated with inflammatory grade and Mallory–Denk bodies on liver biopsy in patients with alcoholic hepatitis [48]. Whether Penicillium plays a role in the pathogenesis of ALD remains to be studied in the future.
Mycobiome in obesity and nonalcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease worldwide with around 30% of the entire population being affected [87]. NAFLD is associated with obesity and often considered the liver manifestation of the metabolic syndrome, and many factors are involved in the development of this complex disease, such as over-nutrition, genetic, and environmental factors [88, 89]. The spectrum of NAFLD ranges from simple steatosis to nonalcoholic steatohepatitis (NASH), which can progress to cirrhosis and even hepatocellular carcinoma (HCC) [88]. Dysregulation of gut microbiome has been linked to the pathogenesis of NAFLD in animal models and human studies [29, 90, 91]. There are limited studies on the relationship between NAFLD and gut mycobiome. One study used ITS sequencing to evaluate fungal diversity and composition in fecal samples of obese and non-obese subjects. Obese patients exhibited lower fungal diversity at family level, while there was no difference at other taxonomic levels.
Importantly, obese patients have distinct fungal composition compared with non-obese subjects. For example, obese subjects have an increased presence of phylum Ascomycota, class Saccharomyces, and families Dipodascaceae and Saccharomycetaceae and, an increased relative abundance of class Tremellomycetes compared with non-obese subjects. In addition, fecal samples of obese subjects were most abundant in Candida, Nakaseomyces, and Penicillium genera, while Mucor was the most prevalent genus in non-obese subjects. Furthermore, the decreased relative abundance of Mucor genus in obese subjects was reversible after weight loss [50]. Whether changes in mycobiota increase the risk or even contribute to obesity development is not known. Another study characterized gut fungal changes using ITS2 sequencing in mice fed with high-fat diet. Mice fed with high-fat diet exhibited similar fungal diversity compared with standard chow diet. The abundance of Alternaria, Saccharomyces, Septoriella, and Tilletiopsis genera; Saccharomyces cerevisiae; and Tilletiopsis washingtonensis were higher in mice fed with standard chow diet [92]. Notably, the predominant colonizer and pathobiont C. albicans were detected in both groups with similar relative abundance. Strong diet-specific coabundance relationships were observed between bacteria and fungi, and the number of coabundance correlations was reduced in high-fat diet-fed mice.
The probiotic Saccharomyces boulardii (S. boulardii) has been widely used as probiotic in clinical practice. It functions by consuming oxygen, which produces an anaerobic environment and facilitates the growth of bifidobacterial and lactic acid bacteria while inhibiting pathogens [93, 94]. In mice with diet-induced diabetes and obesity, oral gavage of S. boulardii daily for 4 weeks reduced body weight, fat mass, hepatic steatosis, and inflammation. S. boulardii increased cecum weight and induced gut microbiota composition changes at the phylum, family, and genus levels [95]. Similarly, in a rat model of diet-induced steatohepatitis, oral gavage of S. boulardii (7.5X109 CFU/kg/d) for 8 weeks reduced body weight, hepatic steatosis, endotoxemia, and inflammation. It also regulates the ratio of Escherichia coli/Bacteroides in the intestine of rats, indicating a potential interaction between bacteria and fungi [96]. It is currently unknown whether fungi contribute to NAFLD pathogenesis, which requires further research in the future.
Mycobiome and primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterized by the development of biliary inflammation and fibro-obliterative lesions [97, 98]. Previous studies have implicated the role of gut microbiota and its metabolites in the pathogenesis of PSC and PSC-associated inflammatory bowel disease (IBD), which exists in the majority of PSC patients [99]. Although PSC is considered a rare disease in global population, it is associated with an increased risk of colorectal and hepatobiliary malignancy, even cholangiocarcinoma [100]. The gut microbiota and its metabolites have been implicated in the pathogenesis of PSC and PSC/IBD [101] as well as with disease severity and clinical outcome [102]. Studies on gut microbiome showed enriched level of Veillonella, Streptococcus, and Enterococcus in PSC patients [103]. Among these altered taxa, Veillonella spp. can produce amino oxidases such as vascular adhesion protein-1 (VAP-1) that facilitates adhesion of gut-trophic lymphocytes to liver endothelium in a substrate-dependent manner. Elevated level of circulating form of VAP-1 is increased in PSC patients, and it is associated with disease severity and clinical outcome [102]. As most studies focus on the gut bacteria in PSC pathogenesis, the role of fungi has not been extensively studied. A possible role of fungi is first implicated by a high prevalence of serum ASCA in PSC patients [104]. Furthermore, Candida spp. (albicans, glabrata, tropicalis, and undifferentiated) in bile identified by culture-based methods is associated with a poor prognosis in patients with PSC, and these patients need liver transplantation relatively soon [105]. Recently, a study investigated fungal changes in feces from patients with PSC changes by ITS2 sequencing. Interestingly, alpha diversity (Shannon and Chao1 indexes) showed no differences among healthy subjects, patients with IBD only, PSC and IBD, and PSC only. Beta diversity analysis revealed a clustering of samples according to the groups. Furthermore, patients with PSC had a higher ITS2/16S diversity ratio compared with IBD only patients, suggesting an increased fungi-to-bacteria diversity ratio. Overall, the fungal microbiota was dominated by two phyla: Ascomycota and Basidiomycota. PSC patients had increased proportions of Exophiala genus and Sordariomycetes class, while the proportion of Saccharomycetaceae family and Saccharomyces cerevisiae species were decreased [97]. In particular, Saccharomyces cerevisiae was also decreased in patients with active IBD, and it is shown to have anti-inflammatory effects by producing cytokine interleukin (IL) 10 [45]. Exophiala genus is involved in infections known as phaeohyphomycosis in immunocompromised hosts. One case report showed that Exophiala dermatitidis caused systemic infection mimicking PSC in a patient without immunodeficiency [106]. Another case report found that Exophiala dermatitidis infection leads to liver cirrhosis and intrahepatic bile duct dilation [107]. The above studies suggest an altered fungal composition with an altered fungi–bacteria correlation network in PSC [97]. Future studies are needed to elucidate the mechanistic role of fungi on PSC development, and in particular, whether fungi contribute to intestinal inflammation. A deeper understanding might pave the way for developing mycobiome-based biomarkers and therapeutic targets in patients with PSC.
Mycobiome in cirrhosis and hepatocellular carcinoma
Cirrhosis is a leading cause of death worldwide resulting from chronic liver diseases such as ALD, viral hepatitis, and NAFLD [108]. One major reason for the mortality is related to gut originated infections [109]. Gut bacteria have been characterized in fecal samples from cirrhosis patients by 16S rDNA sequencing. In particular, Bacteroides genus was dominant in both cirrhosis and healthy groups, but the level was significantly decreased in cirrhosis group. Veillonella, Streptococcus, Clostridium, and Prevotella were enriched in cirrhosis group, while Eubacterium and Alistipes were dominant in healthy groups [110, 111]. While cirrhosis is associated with infections, the role of fungi has been increasingly recognized. One study including 143 cirrhotics (outpatients and inpatients) and 26 controls investigated the bacteria and fungi signature changes and the fungal stability over time in response to antibiotic use. In this study, they found Bacteroidetes/Ascomycota ratio changes with cirrhosis severity. Those changes also can predict 90-day hospitalizations in patients with cirrhosis independent of disease severity, encephalopathy, and clinical biomarkers. Fungal diversity parallels bacterial diversity, and is decreased in cirrhosis compared with healthy controls and negatively correlated with model for end-stage liver disease (MELD) score. Antibiotic therapy reduced bacterial and fungal diversity in outpatients with cirrhosis, while proton pump inhibitor use did not affect fungal diversity [51]. Another study demonstrated that patients with cirrhosis secondary to chronic hepatitis B infection have increased fungal abundance and higher fungal diversity compared with patients with chronic hepatitis B, carriers, and healthy controls, suggesting a positive correlation between fungal diversity and disease severity [112].
Hepatocellular carcinoma (HCC) is the most common primary cancer of the liver and is the sixth most common cancer in western countries [113]. Previous studies showed that fecal bacterial diversity was increased from patients with cirrhosis to early HCC with cirrhosis, both of which were caused by chronic HBV infection [114]. By comparing NASH-induced cirrhosis with or without HCC, patients with HCC had increased level of Bacteroides and Ruminococcaceae, whereas Akkermansia and Bifidobacterium were decreased [115]. The role of gut mycobiome is being less recognized and mainly focused on the role of aflatoxin, a food contaminant and carcinogen produced by fungi (such as Aspergillus flavus and Aspergillus parasiticus), in the pathogenesis of HCC [116]. Aflatoxin B1 is the most abundant member of the family, and it has been shown to contribute to HCC development by being converted to the reactive intermediate AFB1-8, 9 epoxide (AFBO) through cytochrome–P450 enzymes, that binds to hepatocellular DNA [117, 118]. The resulting DNA adducts interact with guanine bases of DNA and cause mutation in the p53 tumor suppressor gene that leads to HCC [118]. HBV and aflatoxin B1 are known to co-exist in countries with high incidence of HCC, suggesting a possible interaction between the two factors [119]. HBV infection may sensitize hepatocytes to the carcinogenic effects of Aflatoxin B1 directly or indirectly, but the exact mechanism is unknown [120].
Fungi–bacteria interactions in human disease
Currently, fungi–bacteria interactions have gained attention due to their impact on human health. Most studies have been focusing on the interactions between Candida species and bacterial species, since they are the most common infectious species in humans, and C. albicans being the leading pathogenic species out of over 150 known species [121]. In the human gastrointestinal tract, the interaction between C. albicans and the Gram-positive bacterium Enterococcus faecalis (E. faecalis) promotes a synergistic and non-pathogenic association with the host [122]. Interestingly, previous studies found increased abundance of C. albicans and E. faecalis in patients with alcoholic hepatitis compared with alcohol use disorder and non-alcoholic controls [67, 83]. If interactions really drive the disease progression or are the result of chronic disease remains to be investigated. In addition, recent studies in murine model demonstrate the existence of interaction between C. albicans and Clostridium difficile (C. difficile) in the context of C. difficile infection. In particular, mice pre-colonized with C. albicans showed increased survival and expression of Il17a in colon in C. difficile infection. Although there was no difference in C. difficile spore and toxin production, the beneficial bacterial genera Bifidobacterium and Akkermansia were increased in C. albicans pre-colonized mice [123]. Therefore, the interactions between bacteria and fungi may serve as important targets or new therapeutic tools in the host health and disease.
Conclusions and perspectives
As the impact of human microbiome receives increasing attention, the study of mycobiome in host health and disease is still at an early stage. Although a number of studies have emerged to characterize the fungal signature in animal models and human studies, it remains unclear whether changes in mycobiome are consequences of the disease process or play pathogenic roles in disease etiology. Especially the potential link between changes in the mycobiome and altered immune responses driving inflammatory liver diseases is currently largely unknown and requires further investigation. While we have discussed studies whereby fungi are involved in liver diseases, there is a clear gap in this field. To date, most studies in the field of liver disease research focus on characterizing fungal signatures in patients with ALD, PSC, and cirrhosis. Additional research is needed to understand the complex interactions between fungi and other groups of microorganisms (bacteria and viruses), and how the interactions affect the host. Targeting mycobiome by anti-fungals or by fecal microbiota transplantation would help us manipulate fungi and understand its functionality.
Abbreviations
- ALD:
-
Alcohol-associated liver disease
- AUD:
-
Alcohol use disorder
- NAFLD:
-
Nonalcoholic fatty liver disease
- NASH:
-
Nonalcoholic steatohepatitis
- PSC:
-
Primary sclerosing cholangitis
- HCC:
-
Hepatocellular carcinoma
- ITS:
-
Internal transcribed spacer
- PCR:
-
Polymerase chain reaction
- AhR:
-
Aryl hydrocarbon receptor
- CD:
-
Crohn’s disease
- PAMP:
-
Pathogen-associated-molecular-pattern
- ASCA:
-
Anti-Saccharomyces cerevisiae antibodies
- S. cerevisiae :
-
Saccharomyces cerevisiae
- CLEC7A:
-
C-type lectin-like receptor 7a
- ECE1 :
-
Extent of cell elongation 1
- S. boulardii :
-
Saccharomyces boulardii
- VAP-1:
-
Vascular adhesion protein-1
- IL:
-
Interleukin
- IBD:
-
Inflammatory bowel disease
- MELD:
-
Model for end-stage liver disease
- HBV:
-
Hepatitis B virus
- E. faecalis :
-
Enterococcus faecalis
- C. difficile :
-
Clostridium difficile
References
Clemente JC, Ursell LK, Parfrey LW, et al. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–70.
Huseyin CE, O’Toole PW, Cotter PD, et al. Forgotten fungi-the gut mycobiome in human health and disease. FEMS Microbiol Rev. 2017;41:479–511.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23.
Lai GC, Tan TG, Pavelka N. The mammalian mycobiome: a complex system in a dynamic relationship with the host. Wiley Interdiscip Rev Syst Biol Med. 2019;11:e1438.
Hernández-Santos N, Klein BS. Through the scope darkly: the gut mycobiome comes into focus. Cell Host Microbe. 2017;22:728–9.
Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Richard ML, Sokol H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2019;16:331–45.
Seed PC. The human mycobiome. Cold Spring Harb Perspect Med. 2014;5:a019810.
Redecker D, Kodner R, Graham LE. Glomalean fungi from the Ordovician. Science. 2000;289:1920–1.
Blackwell M. The fungi: 1, 2, 3… 5.1 million species? Am J Bot. 2011;98:426–38.
Zolan ME. Chromosome-length polymorphism in fungi. Microbiol Rev. 1995;59:686–98.
Nash AK, Auchtung TA, Wong MC, et al. The gut mycobiome of the human microbiome project healthy cohort. Microbiome. 2017;5:153.
Dupuy AK, David MS, Li L, et al. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: discovery of Malassezia as a prominent commensal. PLoS ONE. 2014;9:e90899.
Zhang E, Tanaka T, Tajima M, et al. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol Immunol. 2011;55:625–32.
Huseyin CE, Rubio RC, O’Sullivan O, et al. The fungal frontier: a comparative analysis of methods used in the study of the human gut mycobiome. Front Microbiol. 2017;8:1432.
Nguyen LD, Viscogliosi E, Delhaes L. The lung mycobiome: an emerging field of the human respiratory microbiome. Front Microbiol. 2015;6:89.
Ghannoum MA, Jurevic RJ, Mukherjee PK, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6:e1000713.
Peters BA, Wu J, Hayes RB, et al. The oral fungal mycobiome: characteristics and relation to periodontitis in a pilot study. BMC Microbiol. 2017;17:157.
Burge HA. An update on pollen and fungal spore aerobiology. J Allergy Clin Immunol. 2002;110:544–52.
Charlson ES, Diamond JM, Bittinger K, et al. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med. 2012;186:536–45.
Hoffmann C, Dollive S, Grunberg S, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE. 2013;8:e66019.
Gaitanis G, Magiatis P, Hantschke M, et al. The Malassezia genus in skin and systemic diseases. Clin Microbiol Rev. 2012;25:106–41.
Jo JH, Deming C, Kennedy EA, et al. Diverse human skin fungal communities in children converge in adulthood. J Invest Dermatol. 2016;136:2356–63.
Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(Suppl 4):2–15.
Brown GD, Denning DW, Gow NA, et al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4(165):rv13.
Li XV, Leonardi I, Iliev ID. Gut mycobiota in immunity and inflammatory disease. Immunity. 2019;50:1365–79.
Bacher P, Hohnstein T, Beerbaum E, et al. Human anti-fungal Th17 Immunity and pathology rely on cross-reactivity against candida albicans. Cell. 2019;176(1340–55):e15.
Sparber F, De Gregorio C, Steckholzer S, et al. the skin commensal yeast malassezia triggers a type 17 response that coordinates anti-fungal immunity and exacerbates skin inflammation. Cell Host Microbe. 2019;25(389–403):e6.
Jiang L, Schnabl B. Gut microbiota in liver disease: what do we know and what do we not know? Physiol (Bethesda). 2020;35:261–74.
Thomas S, Izard J, Walsh E, et al. The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res. 2017;77:1783–812.
Foster KR, Schluter J, Coyte KZ, et al. The evolution of the host microbiome as an ecosystem on a leash. Nature. 2017;548:43–51.
Forbes JD, Bernstein CN, Tremlett H, et al. A fungal world: could the gut mycobiome be involved in neurological disease? Front Microbiol. 2018;9:3249.
Iliev ID, Leonardi I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat Rev Immunol. 2017;17:635–46.
van de Veerdonk FL, Netea MG. T-cell subsets and antifungal host defenses. Curr Fungal Infect Rep. 2010;4:238–43.
Scheffold A, Bacher P. Anti-fungal T cell responses in the lung and modulation by the gut-lung axis. Curr Opin Microbiol. 2020;56:67–73.
Vinh DC. Insights into human antifungal immunity from primary immunodeficiencies. Lancet Infect Dis. 2011;11:780–92.
Mengesha BG, Conti HR. The role of IL-17 in protection against mucosal candida infections. J Fungi (Basel). 2017;3(4):52.
Li J, Casanova JL, Puel A. Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol. 2018;11:581–9.
Corvilain E, Casanova JL, Puel A. Inherited CARD9 deficiency: invasive disease caused by ascomycete fungi in previously healthy children and adults. J Clin Immunol. 2018;38:656–93.
Atarashi K, Tanoue T, Ando M, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015;163:367–80.
Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46.
Shao TY, Ang WXG, Jiang TT, et al. Commensal candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe. 2019;25(404–17):e6.
Bacher P, Heinrich F, Stervbo U, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell. 2016;167(1067–78):e16.
Rizzetto L, Kuka M, De Filippo C, et al. Differential IL-17 production and mannan recognition contribute to fungal pathogenicity and commensalism. J Immunol. 2010;184:4258–68.
Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–48.
Standaert-Vitse A, Sendid B, Joossens M, et al. Candida albicans colonization and ASCA in familial Crohn’s disease. Am J Gastroenterol. 2009;104:1745–53.
Limon JJ, Tang J, Li D, et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25(377–88):e6.
Lang S, Duan Y, Liu J, et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology. 2020;71:522–38.
Yang AM, Inamine T, Hochrath K, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest. 2017;127:2829–41.
Mar Rodriguez M, Perez D, Javier Chaves F, et al. Obesity changes the human gut mycobiome. Sci Rep. 2015;5:14600.
Bajaj JS, Liu EJ, Kheradman R, et al. Fungal dysbiosis in cirrhosis. Gut. 2018;67:1146–54.
Wheeler ML, Limon JJ, Bar AS, et al. Immunological consequences of intestinal fungal dysbiosis. Cell Host Microbe. 2016;19:865–73.
Sovran B, Planchais J, Jegou S, et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome. 2018;6:152.
Jostins L, Ripke S, Weersma RK, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24.
Standaert-Vitse A, Jouault T, Vandewalle P, et al. Candida albicans is an immunogen for anti-Saccharomyces cerevisiae antibody markers of Crohn’s disease. Gastroenterology. 2006;130:1764–75.
Rau M, Schilling AK, Meertens J, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. 2016;196:97–105.
Gomes AL, Teijeiro A, Buren S, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2016;30:161–75.
Parfieniuk-Kowerda A, Swiderska M, Szulzyk T, et al. Serum concentrations of Th17-associated interleukins and autoimmune phenomena are associated with the degree of liver damage in alcoholic liver disease. J Gastrointestin Liver Dis. 2017;26:269–74.
Chu S, Sun R, Gu X, et al. Inhibition of sphingosine-1-phosphate-induced Th17 cells ameliorates alcoholic steatohepatitis in mice. Hepatology. 2020;. https://doi.org/10.1002/hep.31321.
Chiaro TR, Soto R, Zac Stephens W, et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci Transl Med. 2017;9(380):eaaf9044.
Sparber F, LeibundGut-Landmann S. Host responses to Malassezia spp. in the mammalian skin. Front Immunol. 2017;8:1614.
Napolitano M, Patruno C. Aryl hydrocarbon receptor (AhR) a possible target for the treatment of skin disease. Med Hypotheses. 2018;116:96–100.
Morris AJ, Byrne TC, Madden JF, et al. Duration of incubation of fungal cultures. J Clin Microbiol. 1996;34:1583–5.
Scanlan PD, Marchesi JR. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J. 2008;2:1183–93.
Hamad I, Sokhna C, Raoult D, et al. Molecular detection of eukaryotes in a single human stool sample from Senegal. PLoS ONE. 2012;7:e40888.
Gouba N, Raoult D, Drancourt M. Plant and fungal diversity in gut microbiota as revealed by molecular and culture investigations. PLoS ONE. 2013;8:e59474.
Chu H, Duan Y, Lang S, et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J Hepatol. 2020;72:391–400.
Pfaller MA, Houston A, Coffmann S. Application of CHROMagar Candida for rapid screening of clinical specimens for Candida albicans, Candida tropicalis, Candida krusei, and Candida (Torulopsis) glabrata. J Clin Microbiol. 1996;34:58–61.
Kaneko T, Makimura K, Onozaki M, et al. Vital growth factors of Malassezia species on modified CHROMagar Candida. Med Mycol. 2005;43:699–704.
Hamad I, Ranque S, Azhar EI, et al. Culturomics and amplicon-based metagenomic approaches for the study of fungal population in human gut microbiota. Sci Rep. 2017;7:16788.
Anderson IC, Cairney JW. Diversity and ecology of soil fungal communities: increased understanding through the application of molecular techniques. Environ Microbiol. 2004;6:769–79.
Iwen PC, Hinrichs SH, Rupp ME. Utilization of the internal transcribed spacer regions as molecular targets to detect and identify human fungal pathogens. Med Mycol. 2002;40:87–109.
Schoch CL, Seifert KA, Huhndorf S, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 2012;109:6241–6.
Tang J, Iliev ID, Brown J, et al. Mycobiome: approaches to analysis of intestinal fungi. J Immunol Methods. 2015;421:112–21.
Nilsson RH, Ryberg M, Kristiansson E, et al. Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS ONE. 2006;1:e59.
Koljalg U, Larsson KH, Abarenkov K, et al. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005;166:1063–8.
Findley K, Oh J, Yang J, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–70.
Schoch CL, Robbertse B, Robert V, et al. Finding needles in haystacks: linking scientific names, reference specimens and molecular data for fungi. Database (Oxford). 2014;2014:bau061
Mathurin P, Lucey MR. Management of alcoholic hepatitis. J Hepatol. 2012;56(Suppl 1):S39-45.
Crabb DW, Im GY, Szabo G, et al. Diagnosis and treatment of alcohol-associated liver diseases: 2019 practice guidance from the American association for the study of liver diseases. Hepatology. 2020;71:306–33.
Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513–24.
Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148:30–6.
Duan Y, Llorente C, Lang S, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11.
Bull-Otterson L, Feng W, Kirpich I, et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE. 2013;8:e53028.
Jiang L, Lang S, Duan Y, et al. Intestinal virome in patients with alcoholic hepatitis. Hepatology. 2020. https://doi.org/10.1002/hep.31459.
Maccioni L, Gao B, Leclercq S, et al. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes. 2020;12:1782157.
Estes C, Razavi H, Loomba R, et al. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123–33.
Brunt EM, Wong VW, Nobili V, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. 2015;1:15080.
Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol. 2017;23:1–12.
Leung C, Rivera L, Furness JB, et al. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. 2016;13:412–25.
Boursier J, Mueller O, Barret M, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764–75.
Heisel T, Montassier E, Johnson A, et al. High-fat diet changes fungal microbiomes and interkingdom relationships in the murine gut. mSphere. 2017;. https://doi.org/10.1128/mSphere.00351-17.
You N, Zhuo L, Zhou J, et al. the role of intestinal fungi and its metabolites in chronic liver diseases. Gut Liver. 2020;14:291–6.
Kelesidis T, Pothoulakis C. Efficacy and safety of the probiotic Saccharomyces boulardii for the prevention and therapy of gastrointestinal disorders. Therap Adv Gastroenterol. 2012;5:111–25.
Everard A, Matamoros S, Geurts L, et al. Saccharomyces boulardii administration changes gut microbiota and reduces hepatic steatosis, low-grade inflammation, and fat mass in obese and type 2 diabetic db/db mice. mBio. 2014;5:e11011–4.
Liu YT, Li YQ. Wang YZ [Protective effect of Saccharomyces boulardii against intestinal mucosal barrier injury in rats with nonalcoholic fatty liver disease]. Zhonghua Gan Zang Bing Za Zhi. 2016;24:921–6.
Lemoinne S, Kemgang A, Ben Belkacem K, et al. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut. 2020;69:92–102.
Dyson JK, Beuers U, Jones DEJ, et al. Primary sclerosing cholangitis. Lancet. 2018;391:2547–59.
Shah A, Macdonald GA, Morrison M, et al. Targeting the gut microbiome as a treatment for primary sclerosing cholangitis: a conceptional framework. Off J Am Coll Gastroenterol. 2020;115(6):814–22.
Fung BM, Lindor KD, Tabibian JH. Cancer risk in primary sclerosing cholangitis: epidemiology, prevention, and surveillance strategies. World J Gastroenterol. 2019;25:659–71.
Little R, Wine E, Kamath BM, et al. Gut microbiome in primary sclerosing cholangitis: a review. World J Gastroenterol. 2020;26:2768–80.
Trivedi PJ, Tickle J, Vesterhus MN, et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut. 2018;67:1135–45.
Ruhlemann M, Liwinski T, Heinsen FA, et al. Consistent alterations in faecal microbiomes of patients with primary sclerosing cholangitis independent of associated colitis. Aliment Pharmacol Ther. 2019;50:580–9.
Muratori P, Muratori L, Guidi M, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA) and autoimmune liver diseases. Clin Exp Immunol. 2003;132:473–6.
Rudolph G, Gotthardt D, Kloters-Plachky P, et al. Influence of dominant bile duct stenoses and biliary infections on outcome in primary sclerosing cholangitis. J Hepatol. 2009;51:149–55.
Oztas E, Odemis B, Kekilli M, et al. Systemic phaeohyphomycosis resembling primary sclerosing cholangitis caused by Exophiala dermatitidis. J Med Microbiol. 2009;58:1243–6.
Hong KH, Kim JW, Jang SJ, et al. Liver cirrhosis caused by Exophiala dermatitidis. J Med Microbiol. 2009;58:674–7.
El Bialy SA, El Kader KF, El-Ashmawy MB. Current progress in antifibrotics. Curr Med Chem. 2011;18:3082–92.
Tandon P, Garcia-Tsao G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin Liver Dis. 2008;28:26–42.
Chen Y, Yang F, Lu H, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–72.
Qin N, Yang F, Li A, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64.
Chen Y, Chen Z, Guo R, et al. Correlation between gastrointestinal fungi and varying degrees of chronic hepatitis B virus infection. Diagn Microbiol Infect Dis. 2011;70:492–8.
Choo SP, Tan WL, Goh BKP, et al. Comparison of hepatocellular carcinoma in Eastern versus Western populations. Cancer. 2016;122:3430–46.
Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014.
Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. 2019;69:107–20.
Kew MC. Aflatoxins as a cause of hepatocellular carcinoma. J Gastrointestin Liver Dis. 2013;22:305–10.
Lopez C, Ramos L, Bulacio L, et al. Aflatoxin B1 content in patients with hepatic diseases. Med (B Aires). 2002;62:313–6.
Hamid AS, Tesfamariam IG, Zhang Y, et al. Aflatoxin B1-induced hepatocellular carcinoma in developing countries: geographical distribution, mechanism of action and prevention. Oncol Lett. 2013;5:1087–92.
Kirk GD, Lesi OA, Mendy M, et al. 249(ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene. 2005;24:5858–67.
Kew MC. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003;23:405–9.
Allison DL, Willems HME, Jayatilake J, et al. Candida-bacteria interactions: their impact on human disease. Microbiol Spectr. 2016;. https://doi.org/10.1128/microbiolspec.VMBF-0030-2016.
Garsin DA, Lorenz MC. Candida albicans and Enterococcus faecalis in the gut: synergy in commensalism? Gut Microbes. 2013;4:409–15.
Markey L, Shaban L, Green ER, et al. Pre-colonization with the commensal fungus Candida albicans reduces murine susceptibility to Clostridium difficile infection. Gut Microbes. 2018;9:497–509.
Acknowledgements
This study was supported in part by a Biocodex Microbiota Foundation Grant, NIH grants R01 AA24726, R01AA020703, U01 AA026939, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (to B.S.) and services provided by P30 DK120515 and P50 AA011999 and by grants from Fond National de Recherche Scientifique Belgium (J.0146.17 and T.0217.18) and Action de Recherche Concertée (ARC), Université Catholique de Louvain, Belgium to P.S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
B.S. has been consulting for Ferring Research Institute, HOST Therabiomics, Intercept Pharmaceuticals, Mabwell Therapeutics, and Patara Pharmaceuticals. B.S.’s institution UC San Diego has received grant support from Axial Biotherapeutics, BiomX, CymaBay Therapeutics, NGM Biopharmaceuticals, and Synlogic Operating Company.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, L., Stärkel, P., Fan, JG. et al. The gut mycobiome: a novel player in chronic liver diseases. J Gastroenterol 56, 1–11 (2021). https://doi.org/10.1007/s00535-020-01740-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-020-01740-5