
Abstract
The intestinal tract is one of the most complex organs of the human body. It has to exercise various functions including food and water absorption, as well as barrier and immune regulation. These functions affect not only the gut itself, but influence the overall health of the organism. Diseases involving the gastrointestinal tract such as inflammatory bowel disease and colorectal cancer therefore severely affect the patient's quality of life and can become life-threatening. Intestinal epithelial cells (IECs) play an important role in intestinal inflammation, infection, and cancer development. IECs not only constitute the first barrier in the gut against the lumen, they also constantly signal information about the gut lumen to immune cells, thereby influencing their behaviour. In contrast, by producing various antimicrobial peptides, IECs shape the microbial community within the gut. IECs also respond to cytokines and other mediators of immune cells in the lamina propria. Interactions between epithelial cells and immune cells in the intestine are responsible for gut homeostasis, and modulations of this crosstalk have been reported in studies of gut diseases. This review discusses the wide field of immune-epithelial interactions and shows the importance of immune-epithelial crosstalk in the intestine to gut homeostasis and the overall health status.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The intestinal epithelium in gastrointestinal diseases
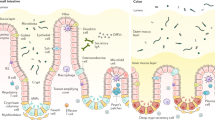
Infection, inflammation, and tumour development are common diseases of the human gut and pose serious threats to health and life. The gut is a biological system of very high complexity and is considered to be the largest immunological organ in the human organism, since it harbours an estimated 80 % of the immune cells of the body [1]. Immune cells and intestinal epithelial cells (IECs) represent the major components of the gut. The intestinal epithelium is composed of a single cell layer that constitutes the lining of the gut, the outer barrier against the lumen. But the thin epithelial cell layer is much more than just a simple barrier that is crucial for the absorption of nutrients and recovery of water. Recent studies have highlighted the role of IECs in the regulation and adaption of mucosal immune responses. The intestinal epithelium undergoes continuous and rapid self-renewal. Most IECs have a very short life span since they are renewed every 4–5 days [2]. This self-renewal process needs to be tightly regulated, and irregularities might cause pathologies [3]. The intestinal epithelium is composed of several mature cell types, such as enterocytes, enteroendocrine cells, goblet cells, and Paneth cells [2]. These cell types are functionally different and essential to fulfill all the different tasks of the intestinal epithelium. For example, enteroendocrine cells secrete hormones such as secretin and gastrin, thereby influencing other organs like the pancreas and stomach [4]. IECs not only produce substances acting on distant organs, but also constantly interact with subjacent immune cells, thereby ensuring the maintenance of a healthy gastrointestinal tract. Disruptions of this complex system are involved in many diseases such as chronic infections, inflammatory bowel disease (IBD), and colon cancer.
Pathogenic bacteria can overgrow the commensal flora in the gut and cause severe diseases, often including diarrhoea. An estimated 2.5 million deaths per year are the consequence of infections of the gastrointestinal tract accompanied by diarrhoea and dehydration [5]. For example, pathogenic Escherichia coli such as enteropathogenic Escherichia coli (EPEC) and enterohaemorrhagic Escherichia coli (EHEC) are key causes of diarrhoeal diseases [6, 7]. Infections with EPEC cause non-specific gastroenteritis, low-grade fever, vomiting, and diarrhoea [8]. EHEC infections lead to severe, crampy abdominal pain, bloody diarrhoea, and can cause haemolytic uraemic syndrome (HUS) [9]. Severe infections from enterohaemorrhagic E. coli occurred in spring 2011 in Germany, with an excess of 2,900 cases of acute gastroenteritis, more than 850 cases of HUS, and at least 50 EHEC-related deaths [10]. IECs are of utmost importance to maintain intestinal homeostasis and to combat intestinal infections. The intestinal epithelium not only represents a physical barrier protecting against the invasion of pathogens, it also senses information from microbial communities in the gut lumen and signals this information to the subjacent immune cells [11]. Most importantly, IECs possess antibacterial properties essential for host defence against pathogens: they secrete antimicrobial peptides (AMPs) into the gut lumen and therefore contribute to the elimination of bacterial pathogens [12]. Moreover, the regulated expression of many such antimicrobial peptides is considered to shape the gut flora in an efficient way to allow the commensal flora to occupy niches that could otherwise be exploited by pathogenic bacteria [13]. In that way, the intestinal epithelium contributes to gut homeostasis.
Other good examples for the important function of the intestinal epithelium are inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis [14]. Inflammatory bowel diseases are described by a chronic inflammation of the gut mucosa characterized by intestinal epithelial damage and infiltration of immune cells. Despite considerable research efforts worldwide, the pathogenesis of IBD is still incompletely understood and the present therapy is not curative, but merely symptomatic. A variety of different factors are involved in the aetiology of IBD such as genetic and environmental factors, deregulated immune responses, and the bacterial microflora. Although the exact aetiology is not known, research on animal models and genome-wide association studies have made it evident that defects in the intestinal epithelium may contribute as a major aetiological factor to the pathogenesis of IBD [15, 16]. Accordingly, disruption of the intestinal epithelium or defects in its barrier function may lead to the invasion of components of the commensal microflora. Although commensal bacteria are generally tolerated in the gut lumen, their deregulated invasion into the bowel wall is believed to cause deregulation of the immune response and chronic inflammation.
One mechanism by which the intestinal barrier prevents a deregulated invasion of bacteria into the bowel wall is the establishment of a mucus layer by goblet cells. The mucus physically separates the intestinal epithelium from the luminal contents, thereby limiting the access of bacteria [17]. Disruption of this mucus was demonstrated to cause pathologies. Accordingly, mice deficient for the mucin Muc2, a major component of the mucus layer, spontaneously developed colitis [18]. It was reasoned that the inflammation resulted from the direct contact of bacteria with the intestinal epithelium [17]. Dependent on the composition of the bacterial microflora in the gut, IECs were demonstrated to produce a wide range of cytokines [19], which in turn stimulate the reaction of subjacent immune cells, potentially causing colitis. In line with this model, increased numbers of activated dendritic cells (DCs) and increased amounts of inflammatory cytokines such as IL (Interleukin)-1β and IL-12p40 were detected in the gut of another mouse strain showing a missense mutation in Muc2 (Winnie mice), which also spontaneously developed intestinal inflammation [20]. Winnie mice showed alterations in the composition of different immune cell populations in the gut towards a T helper (Th)1- and Th17-dominated immune response. The altered cytokine profile and immune cell populations in the guts of mice that lack Muc2 demonstrate the impact of mucus secreted by goblet cells on intestinal immune homeostasis.
Paneth cells are another component of the intestinal epithelial barrier essential for intestinal immune homeostasis. They are located at the bottom of the crypts, in close contact to the stem cells, and secrete factors important for stem cell maintenance such as Wnt3, the Notch ligand DII4 (Delta-like ligand), and EGF (epidermal growth factor) [21]. In addition and of prime importance, Paneth cells produce and secrete antimicrobial granules into the crypt lumen. These granules contain IgA (Immunoglobulin A), TNF-α (tumour necrosis factor alpha), CD95 ligand (CD95L) and antimicrobial peptides, such as lysozyme, secretory phospholipase A2 (sPLA2), α-defensins, and HIP/PAP (hepatocarcinoma-intestine-pancreas/pancreatic-associated protein; mouse counterpart is termed RegIIIγ) [22]. Thereby, the factors released by Paneth cells protect intestinal stem cells and are further dispersed in the mucus layer, generating a general antimicrobial milieu close to the epithelium [23].
Similar to mucus produced by goblet cells, most antimicrobial peptides indirectly regulate immune reactions in the gut: they influence and shape the bacterial gut flora, which in turn affects the host immune status. For example, mice with an altered expression of alpha-defensins showed changes in microbiota composition such as the number of segmented filamentous bacteria (SFB) [13]. Importantly, antimicrobial peptides not only shape the commensal microflora, they are also fundamental for an efficient eradication of bacterial pathogens. Studies on NOD2 (nucleotide-binding oligomerisation domain-containing protein 2) have further highlighted the importance of a tightly regulated antibacterial response. NOD2 was the first gene identified whose polymorphisms were associated with an increased risk for the development of Crohn’s disease [24, 25]. NOD2 is highly expressed in the cytoplasm of Paneth cells and its activation by bacterial muramyl dipeptide leads to NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation followed by production of defensins [26]. Interestingly, the polymorphisms of the IBD risk allele NOD2 have been associated with diminished expression of the α-defensins HD5 and HD6 [27]. Mice deficient for NOD2 also showed decreased expression of certain cryptdins and were highly susceptible to oral infection with Listeria monocytogenes [28]. In line with these findings, several other studies linked changes in antimicrobial peptide expression to an altered susceptibility to infections with gut pathogens. For example, mice overexpressing CRAMP (cathelin-related antimicrobial peptide) were protected from an oral challenge with Citrobacter rodentium [29]. In contrast, mice lacking mature cryptdins were highly susceptible to oral infection with Salmonella Typhimurium [30]. Interestingly, altered expression of some AMPs such as β-defensins and the cathelicidin LL-37 (human orthologue to CRAMP) has been found in patients with Crohn’s disease [31].
Crohn’s disease patients have further been demonstrated to show epithelial barrier dysfunction [32]. Since an altered permeability of the gut epithelium has also been detected in some first-degree relatives without symptoms of Crohn’s disease, the maintenance of barrier function was discussed to be an important factor in the pathogenesis of IBD [33]. Tight junction proteins are essential to maintaining epithelial barrier integrity and regulating trans-epithelial permeability. Interestingly, junction proteins such as claudins and the junctional adhesion molecule-A (JAM-A) were demonstrated to show altered expression in patients with IBD [34, 35]. The tight junction permeability can be regulated by multiple factors such as physiological stimuli without giving access for bacteria [36]. However, when tight junctions are not accurately formed, the increased permeability of the intestinal epithelium can become pathologic. In agreement with this hypothesis, mice with loss of JAM-A showed increased susceptibility to DSS (dextran sodium sulphate) and developed a strong colitis, which might be reasoned by the increased intestinal permeability and subsequent barrier dysfunction observed in these mice [33].
Barrier dysfunction cannot only be the consequence of altered tight junction expression, but also of a deregulated cellular homeostasis of the epithelium. Excessive cell death in the epithelium without appropriate replacement has been shown to drive chronic inflammation similar to IBD [3, 37]. While excessive epithelial cell death can drive barrier dysfunction and inflammation, deficient epithelial cell death promotes intestinal cancer. It has been known for several decades that the accumulation of mutations in substantial genes such as proto-oncogenes, DNA-repair genes and tumour suppressor genes in IECs can deregulate epithelial homeostasis and cause the development of colorectal cancer [38]. However, in addition to such cell intrinsic mechanisms, it has recently been appreciated that a tumour is more than the accumulation of mutations in the tumour cell itself. Tumours contain not only neoplastic IECs, but also a large amount of immune cells. To protect the tumour from an anti-tumour immune response, tumour cells enact immune-escape mechanisms by directly influencing the activity of immune cells [39]. Furthermore, the increased risk for the development of colorectal cancer observed in IBD-patients is believed to be due to the inflammatory microenvironment—inflammatory cytokines produced by activated immune cells can serve as growth factors and favour the proliferation of pre-neoplastic epithelial cells [40–42]. Thus, immune-epithelial interactions in cancerogenesis act in both directions. It is therefore essential for the understanding of tumour pathogenesis and for the development of new therapeutic strategies to not only investigate the tumour cell itself, but also to define the complex interplay between immune cells and tumour cells.
Overall, protection of the gut from severe diseases such as infections, IBD, and cancer are of utmost importance for health and survival. The intestine is composed of immune cells, stromal cells, and epithelial cells that constantly interact for the maintenance of a healthy gut. Efforts to understand these interactions have been increased in the last decade and provide promising novel approaches for targeting gut-related diseases. This review focuses on the importance of such epithelial-immune crosstalk in the gut.
Epithelial cells influence immune cells in the gut
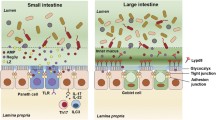
The intestinal epithelium is strategically located between potential antigens within the gut lumen and the large number of immune cells in the lamina propria. IECs not only physically separate these two compartments, they also communicate and translate signals from one compartment to the other. The communication between IECs and immune cells is mediated via cell–cell contact and chemokine and cytokine signalling (Fig. 1). It has been shown that signals from IECs can directly and indirectly act on resident immune cells and thereby shape mucosal immune responses. This communication might by crucial to exerting an appropriate immune response to microbial challenges.

Intestinal epithelial cells have diverse effects on and also respond to factors produced by the subjacent immune cells. The immune-epithelial crosstalk at the intestinal epithelial surface is important for gut homeostasis and during gastrointestinal diseases
IECs induce immune tolerance in the gut
IECs are believed to play a major role in inducing immune tolerance to harmless antigens present in the gut lumen. IECs sense bacteria by means of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), nucleotide-binding site and leucine-rich repeat-containing receptors (NLRs), and retinoic acid inducible gene-I (RIG)-like receptors (RLRs), which detect conserved pathogen-associated molecular patterns (PAMPs) [43–45]. In healthy intestinal tissue, IECs express low levels of TLRs on the apical side and higher levels on the basolateral side [43, 46]. This may also contribute to the tolerance of bacteria in the gut. In fact, activation of TLRs on IECs under steady-state conditions results in the production of factors that are important for homeostasis of the intestinal epithelium [47]. Interestingly, the activation of TLRs on IECs by bacterial products has also been demonstrated to result in the release of the TNF-family member APRIL (a proliferation-inducing ligand), triggering an IgA(2) class switch in B cells [48].
IECs can recognize and take up bacterial antigens [50], process them, and further present them via the major histocompatibility complex (MHC)-class Ib molecule CD1d. This results in the activation of special regulatory T cells and contributes to the tolerogenic milieu of the gut [49, 50]. In addition to the activation of special regulatory T cells, IECs themselves produce suppressive cytokines such as IL-10 that act on the subjacent immune cells [51]. The important role of IL-10 in the lamina propria was established when the investigation of IL-10−/− mice led to the discovery that these mice spontaneously developed chronic enterocolitis [52]. In addition to the induction of regulatory T cells and IL-10 production, IECs were demonstrated to contribute to tolerance by influencing the maturation of tolerogenic dendritic cells. It has recently been shown that antigen-sampling dendritic cells took up Muc2 and that glycans associated with Muc2 imprinted the DCs with anti-inflammatory properties [53]. Furthermore, Rimoldi et al. [54] demonstrated that IECs released TSLP (thymic stromal lymphopoietin), driving dendritic cells towards a tolerogenic phenotype. These tolerogenic DCs suppressed the development of a Th1 response and promoted the polarization towards Th2 cells. An important influence of IECs on DC maturation for immune homeostasis in the gut was further demonstrated by Iliev et al. However, in their study, the release of TGF-β (transforming growth factor beta) and retinoic acid (RA) by IECs, rather than of TSLP, was required for the maturation of tolerogenic DCs, which were able to induce de novo Treg cells [55]. Most interestingly, the transfer of these induced regulatory cells was capable of protecting mice from experimentally-induced colitis, providing evidence that IECs can mediate immune tolerance by directly influencing immune cells. In another study, IECs were shown to regulate DC function via secretion of the integrin αvβ6. Accordingly, the authors showed that uptake of food antigens by IECs upregulated αvβ6 in IECs [56]. Food antigens together with αvβ6 were transported to the extracellular environment via exosomes and uptake of these by dendritic cells resulted in the production of TGF-β and the generation of Tregs. Importantly, IEC-derived signals not only act on lamina propria immune cells, but can influence immune cells at distant sites. For example, exosome-like nanoparticles derived from IECs were shown to carry prostaglandin E2 and migrate to the liver where they act on NKT cells and induce anergy [57].
IECs initiate inflammatory responses
In contrast to the induction of immune tolerance by IECs, several studies have demonstrated an involvement of such an epithelial-immune crosstalk in the induction of gut inflammation.
As mentioned before, IECs express TLRs, allowing them to recognize microbial antigens. However, in contrast to the anti-inflammatory mechanisms described above, under yet-to-be-defined circumstances, TLR-signalling in IECs can promote inflammation. TLR2 and TLR4 expression have been shown to be strongly upregulated on IECs of patients with Crohn’s disease and ulcerative colitis [46], and this might suggest a greater sensitivity of CD and UC patients to the bacterial microflora in the gut [58]. Upon stimulation of IECs via TLR, these cells can produce pro-inflammatory cytokines such as IL-8, IL-18, and TNF-α [59, 60]. Accordingly, infection with pathogenic bacteria such as enterohaemorrhagic Escherichia coli results in the production of the chemokine IL-8 by IECs [61]. IL-8 then recruits immune cells such as neutrophil granulocytes to the site of infections. Interestingly, the capacity of epithelial cells to produce cytokines and chemokines seems to depend on the nature of the signal that IECs are confronted with—while non-invasive bacteria were mostly unable to stimulate epithelial cells for the production of inflammatory cytokines, invasive bacteria stimulated IECs to produce and release pro-inflammatory cytokines and chemokines, which induce the maturation of dendritic cells [62]. For example, human IECs treated with Vibrio cholerae produced the cytokine TSLP and the chemokine CCL20, which attract dendritic cells [63]; Bhowmick et al. [63] demonstrated that the mediators produced by Vibrio cholerae stimulated IECs to activate dendritic cells for the expression and secretion of the inflammatory cytokines TNF-α, IL-6, and IL-1β. Furthermore, the activated dendritic cells now produced the Th2 cell-attracting chemokines CCL17 and CCL22, and promoted differentiation of CD4+ T cells into Th2 cells, producing high amounts of IL-4, IL-13, and TNF-α. In that way, IECs sense pathogenic bacteria and initiate inflammatory responses by influencing the maturation of dendritic cells. Induction of TSLP was also observed for gastric epithelial cells treated with Helicobacter pylori [64].
It has recently been discovered, that some antimicrobial peptides not only show direct antibacterial activities, but also primary innate immune functions, and are involved in the attraction of immune cells. Cathelicidins, such as the human LL-37, which is expressed by surface colonic epithelial cells, was demonstrated to facilitate recruitment of eosinophils and neutrophils [65, 66]. Furthermore, LL-37 induced the expression of chemokines, further supporting the recruitment of immune cells [67]. Although LL-37 is constitutively expressed in the colonic epithelium [66], its expression can be increased under certain conditions such as inflammation and infection [68, 69]. Therefore, it is speculated that the increased LL-37 expression by colonic epithelial cells under certain conditions attracts other immune cells such as neutrophil granulocytes, which have been demonstrated to migrate into the gut lumen during intestinal infections or inflammatory bowel disease [70].
As another example of the direct effect of regulators produced by IECs on immune cells, mice deficient for the secreted goblet cell-derived protein known as resistin-like molecule beta (RELMbeta) failed to develop intestinal inflammation induced by the helminth Trichuris muris [71]. RELMbeta activated macrophages to express MHC class II molecules, to secrete IL-12/23p40, and to mediate antigen-specific IFN-γ (Interferon gamma) expression in CD4+ T cells during Trichuris muris infection. The crosstalk between goblet cells and macrophages seems to be important for the development of a CD4+ T-cell response during infection with parasitic helminths.
IECs promote immunosuppression in tumours
In the human body, neoplastic cells are usually recognized and eliminated by the immune system as soon as they arise. Under certain circumstances, the neoplastic cells are not cleared by the immune system and can give rise to cancer. However, colorectal tumours still harbour immunogenic potential [72, 73]. In response to tumour formation, in several studies it was shown that immune cells infiltrate into the tumour and inhibit tumour growth [74, 75]. These studies showed that the immune system is capable of initiating immune responses to intestinal cancer. However, it is obvious, that the immune system is not always effective in eliminating cancer cells, suggesting that there must be a mechanism allowing the tumours to survive. In fact, human tumours including intestinal cancer are known to develop strategies that allow them to escape surveillance and clearance by the immune system. For example, tumour cells can become resistant to immune reaction or exert immunosuppressive functions [76]. This suppression of the immune system is partially based on a suppression of Th1 responses, which are necessary for the activation of an efficient cytotoxic T-cell response [77]. It has been discussed that colon tumour cells suppress Th1 responses and favour Th2 responses by several different mechanisms, such as Cox-2 (cyclooxygenase-2) and IL-10 overexpression [78]. Cox-2 overexpression was observed in 86 % of colon carcinomas [79]. Cox-2 caused prostaglandin E2 expression, resulting in the reduced activation of NK cells and cytotoxic T lymphocytes and the inhibition of IL-2 production [80]. Colorectal tumour cells have further been shown to produce IL-10 themselves [81]. IL-10 is a pleiotropic cytokine with many immunosuppressive functions and has been shown to be overexpressed in multiple malignant diseases, such as melanomas, squamous carcinoma, renal cell carcinoma and colorectal cancer [78]. Moreover, colon tumour cells have been reported to secrete elevated levels of prostaglandin E2 and TGF-β, resulting in the stimulation of monocytes and intestinal macrophages to produce IL-10 [82]. The secretion of TGF-β by tumour cells in the gut has many additional effects on immune cells in the tumour microenvironment such as the inhibition of T-cell proliferation, differentiation, and maturation, inhibition of the expression of co-stimulatory molecules on dendritic cells, and suppression of macrophage TNF-α, reactive oxygen, and nitric oxide production [78]. Besides the observations that tumour cells favour a Th2 immune response, tumour cells have been reported to secrete VEGF (vascular endothelial growth factor), not only causing neovasculation, but also inhibiting dendritic cell differentiation; the immature dendritic cells in turn inhibited T cell proliferation [83, 84].
Interestingly, colon tumour cells not only influence the subjacent immune cells in regard to their maturation and activation state via the secretion of cytokines: some studies showed that tumour cells in the gut can further express the cell death receptor ligand Fas-ligand (Fas-L), thus triggering lymphocytes to undergo apoptosis [85, 86]. This theory has been termed “tumour counterattack hypothesis” and in that way, the tumour cells themselves escape the immune response by inducing cell death in immune cells. However, this model of tumour immunosuppression is controversial, since other studies showed that colon cancer cells cannot induce T-cell death via Fas-L [87].
Overall, all types of IECs have diverse effects on subjacent immune cells. Mucus produced by goblet cells, Paneth cell products, and the multiple factors released by enterocytes are important for gut homeostasis and are also involved in gut diseases (Fig. 1).
Immune cells influence epithelial cells in the gut
IECs not only produce and release mediators affecting immune cells, they also respond to factors produced by the subjacent immune cells (Fig. 1). That IECs respond to a wide range of cytokines has already been reported two decades ago. IECs were shown to express the common γ chain of the IL-2 receptor, enabling them to interact with IL-2, IL-4, IL-9, and IL-13 [88]. Kumar et al. [89] demonstrated that IL-2 plays an important role in intestinal epithelial homeostasis, since it can induce wound repair mechanisms dependent on Janus kinase 3 (Jak3). The binding of IL-2 to the IL-2 receptor on the IECs was then demonstrated to regulate proliferation and cell death of IECs [90].
Regulation of IECs during intestinal infection and inflammation
Intestinal infections lead to the secretion of a wide range of inflammatory cytokines by lamina propria immune cells, thereby providing constant information about the activation status of the mucosal immune system to the epithelial barrier. For example, bacterial flaggelin has been demonstrated to induce high expression of IL-23 by CD103+CD11b+ dendritic cells present in the lamina propria [91]. This subsequently caused the secretion of IL-22 by immune cells and the induction of IL-22-dependent responses in IECs, such as the expression of the antimicrobial peptide RegIIIγ. Thus, immune cells can regulate the antimicrobial activity of the epithelium. This axis seems to be essential for the efficient fight against pathogenic bacteria, as IL-22-deficient mice infected with Citrobacter rodentium showed decreased RegIIIγ production, increased epithelial damage, systemic bacterial burden and mortality [92]. Immune cells not only induce the production of defence proteins by IECs, they further promote wound healing [93]. During the course of infection, the immune cell-induced elimination of pathogens also affects host tissue and causes tissue destruction. Therefore, effective healing of host tissue is urgently required and managed by the organism in a smart way: cytokines that induce inflammation to efficiently eradicate a pathogen at the same time protect the tissue from the consequences of inflammation by inducing wound healing. A good example is the cytokine IL-6. On the one hand, IL-6 is a pro-inflammatory cytokine influencing immune cells and produced after bacterial infection [92, 94]. On the other hand, IL-6 and also IL-22 at the same time induce Stat3 (signal transducer and activator of transcription 3) signalling in epithelial cells, leading to epithelial regeneration and wound healing [93] (Fig. 2).

Cytokines inducing inflammation at the same time induce healing of damaged intestinal epithelial tissue
During gastrointestinal infections and in the course of Crohn’s disease, highly increased production of the inflammatory cytokine IFN-γ by lymphocytes in the gut has also been observed [95–97]. IECs respond to IFN-γ via the increased expression of MHC I and II and the upregulation of the adhesion molecule ICAM-1 (Intercellular Adhesion Molecule 1) [98]. These changes in the intestinal epithelium are essential to allow antigen-presentation by IECs and adhesion of neutrophils to the intestinal epithelium during infections. Interestingly, the IFN-γ-induced expression of MHC II molecules is blocked by the direct binding of IL-10 to IECs, showing the anti-inflammatory effect of IL-10 also directly on IECs [99]. IFN-γ further contributes to host defence by the modulation of goblet cell function [100]. While INF-γ signalling in lamina propria immune cells was important for the restriction of Salmonella typhimurium, IFN-γ signalling to the intestinal epithelium caused a “loss” of mucus-filled goblet cell vacuoles that was suggested to result from increased mucus secretion [100]. This “loss” of goblet cell vacuoles was not detected in IFN-γ−/− mice infected with S. Typhimurium. The same study demonstrated that IFN-γ−/− mice show increased pathogen colonization of the lamina propria and pathogen loads in the cecal epithelium after S. Typhimurium infection. This might suggest that the reduced goblet cell vacuole secretion in IFN-γ−/− mice resulted in an impaired clearance of bacteria and increased invasion of pathogens. However, counteracting the potential positive effect of IFN-γ on the intestinal epithelial barrier, IFN-γ has been demonstrated to be responsible for the elimination of Paneth cells during infection with parasites [101, 102]. IFN-γ produced by CD4+ Th1 cells during infection with Toxoplasma gondii resulted in Paneth cell death and the loss of antimicrobial peptides [101]. Since Paneth cell loss has also been observed in Crohn’s disease patients, this suggests that IFN-γ might promote intestinal inflammation by inducing Paneth cell death.
In addition to IFN-γ, increased secretion of the inflammatory cytokine TNF-α in the gut has been detected in inflammatory bowel disease patients and was shown to contribute to acceleration of the disease by affecting the intestinal epithelial barrier. TNF-α together with IFN-γ has been shown to alter the permeability of diverse, tight junction proteins via modulation of the actin cytoskeleton or decreased transcription of tight junction genes [103, 104]. As mentioned above, increased permeability of the intestinal epithelial barrier and alterations of epithelial junction adhesion molecule expression have also been described in patients with IBD [35]. It is still not clear whether the increased permeability is causative for the disease or is a consequence of the disease due to the increased amount of inflammatory cytokines. In line with a role for TNF-a in the deregulation of barrier function, it has long been known that anti-TNF-α therapy by antibodies such as adalimumab and infliximab in patients with inflammatory bowel disease can lead to restitution of the intestinal barrier, but the mechanisms involved were largely unknown [105]. Fischer et al. recently demonstrated that adalimumab reversed the downregulatory effects of TNF-α on tight junction proteins, and thereby prevented barrier dysfunction induced by TNF-α [104].
In addition to the modulatory effects of TNF-α on tight junction proteins, TNF-α also has a profound impact on IEC survival. It was recently suggested that cell death triggers are constantly present in the healthy intestine [106]. IECs lacking the crucial apoptosis inhibitor cFlip (Cellular FLICE-Like Inhibitory Protein) died in vivo. Cell death of cFlip-deficient IECs in mice was accompanied by the invasion of inflammatory cells into the gut mucosa [106]. Similarly, another study described epithelial cell death and spontaneous development of inflammatory lesions in the terminal ileum of mice deficient for caspase-8 in the intestinal epithelium [107]. Since cFlip deficiency was associated with increased activation of caspase-8, these studies showed that excessive activation, but also full inhibition of caspase-8 in the intestinal epithelium leads to epithelial cell death, barrier dysfunction, and intestinal inflammation. Interestingly, both studies demonstrated that death of IECs was dependent on extrinsic signals, such as the death receptor ligand TNF-α. These studies also indicated a different susceptibility of IEC types towards cell death. While in cFlip-deficient mice, IECs died from apoptosis independent of their cell type, in caspase-8-deficient mice in the steady state, primarily Paneth cells were affected. The Paneth cell loss observed in caspase-8-deficient mice caused decreased production of AMPs and barrier defects. Similar to caspase-8-deficient mice, Crohn’s disease patients showed epithelial cell death at the crypt base and human Paneth cells were hypersensitive to TNF-induced cell death, demonstrating the potential relevance of immune-epithelial interactions in human diseases [107]. The important effect of TNF-α on the intestinal epithelium was further shown by analysis of mice lacking NF-kappa-B essential modulator (NEMO) in IECs [108]. The inhibition of NF-κB by deletion of NEMO in IECs spontaneously caused severe colitis in these mice. Nenci et al. showed that TNF-α signalling led to apoptosis of NEMO-deficient colonic epithelial cells and impaired expression of AMPs, followed by invasion of bacteria into the mucosa. Overall, TNF-α and other cell death triggers constantly secreted at low levels in the steady state in the gut act on IECs, but execution of cell death is tightly controlled by the intrinsic epithelial cell death machinery.
Immunoregulation of colorectal tumour growth
Interactions between tumour cells and immune cells are more and more becoming the focus of research. However, while the immunosuppression by intestinal tumour cells is of interest for researchers worldwide, as discussed above, the investigation of the influence of immune cells on tumour cells is also of utmost importance. While only about 20 % of colorectal cancers have been linked to heritable genetic changes [109], most colorectal cancers have been associated with environmental influences. For example, patients showing chronic inflammation of the gut mucosa such as IBD patients are at an increased risk for the development of tumours in the gut depending on the duration and extent of inflammation [110]. This observation highlights the important influence of immune cells on IECs and the pathogenesis of colorectal cancer by providing a tumour-promoting inflammatory microenvironment. The presence of inflammatory cells in the mucosa can lead to the secretion of various inflammatory cytokines and the production of reactive oxygen species (ROS) by activated immune cells [111]. ROS can induce DNA damage and mutations in the developing tumour [112]. Another potential mechanism by which immune-epithelial crosstalk might support tumour development is based on the pleiotropic function of some cytokines: IL-1, IL-6, and TNF-α can serve as tumour survival factors and further promote tumour growth [40–42]. The tumour-promoting potential of most inflammatory cytokines might be based on altered homeostatic regulation. Most of the effects of IL-6 on tumour cells are mediated by Stat3. Activation of Stat3 results in proliferation and inhibition of cell death, and therefore contributes to tumour growth: mice deficient for Stat3 in IECs, and therefore also in tumour progenitor cells, were demonstrated to show significantly less and smaller tumours in a colitis-associated tumour model compared to mice expressing normal Stat3 [113]. However, IL-6 and the consequent activation of Stat3 play a dual role in colitis-associated cancer. While the activation of Stat3 promotes tumour growth and survival, the activation of Stat3 in epithelial cells during colitis is protective and necessary for the repair of injured tissue [93]. Another way that Stat3 might promote tumour development is via NF-kB, since Stat3 activation was demonstrated to prolong activation of the transcription factor NF-κB in tumour cells [114].
Another pleiotropic cytokine that is produced by gut immune cells and was demonstrated to have effects on tumour cells is TNF-α. Its tumour-suppressing and tumour-promoting activities have long been discussed, but the mechanisms by which TNF-α affects tumour growth are far from being fully understood [115, 116]. As early as 1975, TNF-α was discovered as a factor produced by human body cells that had the potential to destroy tumours [117]. It was first thought that the destructive effect of TNF-α on tumour cells is dependent on the binding of TNF-α to its receptor expressed on IECs and the subsequent activation of the caspase-cascade, resulting in cell death. It has therefore been hypothesized that the cytokine itself might be a promising cancer treatment. Subsequent studies indeed showed the tumour destructive potential of TNF-α on both xenograft and syngeneic tumours in mice [118, 119]. However, phase I clinical trials turned out to be disappointing: systemic TNF-α administration induced a “cytokine storm” causing severe toxicity and only few tumour responses [120, 121]. It was then reported that a combination of TNF-α, IFN-γ, and melphalan showed higher efficiency in killing tumours [122]. Therefore, the original idea of TNF-α directly killing tumour cells was questioned. It was then hypothesized that TNF-α alone does not have toxic effects on tumour cells and that the earlier reported cytotoxic effect of TNF-α on tumour cell lines was reasoned by the additional presence of metabolic inhibitors such as IFN-γ [115, 123]. In fact, a study from 2010 reported that the blocking of TNF-α in a mouse model of inflammation-associated colon cancer had no detectable effect on tumourigenesis [124]. However, other studies reported that TNF-α can serve as a growth factor at least for some tumours [125, 126]. In line with this observation, Popivanova et al. [127] demonstrated that blocking TNF-α in mice resulted in reduced tumour development in a mouse model of colitis-associated cancer. Since activation of NF-κB is also mediated by TNF-α signalling, TNF-α was thought to act on tumour epithelial cells to promote their proliferation [126]. However, recent publications in 2010 and 2012 again reported TNF-α-induced cell death in colon cancer [128, 129]. Together, although the effect of TNF-α on tumour cells has been extensively studied, its precise and defined actions are still incompletely understood. The contradictory reports of TNF-α activity on tumour cells might be explained by the observation that tumour cells can become resistant to death-receptor ligands such as TNF-α and Fas-L by losing cell surface expression of death receptors [130]. The resistance to death receptor-induced apoptosis was also attributed to altered expression of apoptosis-related molecules such as the apoptosis-inhibitory protein cFlip [131]. In that way, TNF-α might play a dual role in tumour pathogenesis depending on the molecular profile of the tumour cells.
Conclusion
This review highlights the complex interplay at the intestinal epithelial brush border. The intestinal epithelium is centrally located, separating two distinct compartments: the outer environment including bacteria and food antigens, and the inner world, the host tissue. However, the intestinal epithelial barrier does not simply protect the inner world, the organism, from the invasion of components of the outer environment, such as bacteria: increasing evidence indicates that IECs mediate the translation of signals between the microbial world and the host immune system. This interplay not only affects intestinal immune tolerance, but also might be crucial for disease prevention in distant organs. On the other hand, immune cells constantly report the immune status of the gut to the epithelial cells, influencing their behaviour in a way that is appropriate for gut threats such as infections. In the case of diseases such as inflammatory bowel disease or colorectal cancer, these well-balanced interactions are disturbed, and the same mechanisms that are usually protective in the steady state can contribute to IBD or tumour pathogenesis. We are just beginning to understand the complex interplay between the gut flora and the mucosa, as well as the systemic immune system, but it is tempting to speculate that the modulation of epithelial-immune interactions in the gut might be a promising therapeutic strategy for gut-related diseases.
References
Castro GA, Arntzen CJ. Immunophysiology of the gut: a research frontier for integrative studies of the common mucosal immune system. Am J Physiol. 1993;265(4 Pt 1):G599–610.
Van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–60.
Günther C, Neumann H, Neurath MF, et al. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62(7):1062–71.
Ahlman H. Nilsson. The gut as the largest endocrine organ in the body. Ann Oncol. 2001;12(Suppl 2):S63–8.
Kosek M, Bern C, Guerrant RL. The global burden of diarrhoeal disease, as estimated from studies between 1992 and 2000. Bullo World Health Organ. 2003;81(3):197–204.
Clarke SC, Haigh RD, Freestone PP, et al. Virulence of enteropathogenic Escherichia coli, a global pathogen. Clin Microbiol Rev. 2003;16(3):365–78.
Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2(2):123–40.
Donnenberg MS, Whittam TS. Pathogenesis and evolution of virulence in enteropathogenic and enterohaemorrhagic Escherichia coli. J Clin Invest. 2001;107(5):539–48.
Navaneethan U, Giannella RA. Mechanisms of infectious diarrhea. Nat Clin Pract Gastroenterol Hepatol. 2008;5(11):637–47.
Robert Koch Institute. Report: Final presentation and evaluation of epidemiological findings in the EHEC O104:H4 outbreak, Germany 2011. Berlin 2011.
Rescigno M. The intestinal epithelial barrier in the control of homeostasis and immunity. Trends Immunol. 2011;32(6):256–64.
Ayabe T, Ashida T, Kohgo Y, et al. The role of Paneth cells and their antimicrobial peptides in innate host defense. Trends Microbiol. 2004;12(8):394–8.
Salzman NH, Hung K, Haribhai D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11(1):76–83.
Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347(6):417–29.
Pastorelli L, De Salvo C, Mercado JR, et al. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280.
Hamilton MJ, Snapper SC, Blumberg RS. Update on biologic pathways in inflammatory bowel disease and their therapeutic relevance. J Gastroenterol. 2012;47:1–8.
Johansson ME, Phillipson M, Petersson J, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105(39):15064–9.
Van der Sluis M, De Koning BA, De Bruijn AC, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is ciritical for colonic protection. Gastroenterology. 2006;131(1):117–29.
Bahrami B, Macfarlane S, Macfarlane GT. Induction of cytokines formation by human intestinal bacteria in gut epithelial cell lines. J Appl Microbiol. 2011;110(1):353–63.
Eri RD, Adams RJ, Tran TV, et al. An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol. 2011;4(3):354–64.
Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–8.
Ouellette AJ. Paneth cells and innate mucosal immunity. Curr Opin Gastroenterol. 2010;26(6):547–53.
Antoni L, Nuding S, Weller D, et al. Human colonic mucus is a reservoir for antimicrobial peptides. J Crohns Colitis. 2013;. doi:10.1016/j.crohns.2013.05.006.
Hugot JP, Laurent-Puig P, Gower-Rousseau C, et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature. 1996;379(6568):821–3.
Hugot JP, Chamaillard M, Zousali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603.
Voss E, Wehkamp J, Wehkamp K, et al. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem. 2006;281(4):2005–11.
Wehkamp J, Harder J, Weichenthal M, et al. NOD (CARD15) mutations in Crohn’s disease are assocaited with diminished mucsoal alpha-defensin expression. Gut. 2004;53(11):1658–64.
Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–4.
Iimura M, Gallo RL, Hase K, et al. Cathelicidin mediates innate intestial defense against colonization with epithelial adherent bacterial pathogens. J Immunol. 2005;174(8):4901–7.
Wilson CL, Ouellette AJ, Satchell DP, et al. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286(5437):113–7.
Wehkamp J, Schmid M, Stange EF. Defensins and other antimicrobial peptides in inflammatory bowel disease. Curr Opin Gastroenterol. 2007;23(4):370–8.
Silva MA. Intestinal dendritic cells and epithelial barrier dysfunction in Crohn’s disease. Inflamm Bowel Dis. 2009;15(3):436–53.
Katz KD, Hollander D, Vadheim CM, et al. Intestinal permeability in patients with Crohn’s disease and their healthy relatives. Gastroenterology. 1989;97(4):927–31.
Prasad S, Mingrino R, Kaukinen K, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85(9):1139–62.
Vetrano S, Rescigno M, Cera MR, et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135(1):173–84.
Turner JR, Rill BK, Carlson D, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol Cell Physiol. 1997;273:C1378–85.
Chen L, Park SM, Turner JR, et al. Cell death in the colonic epithelium during inflammatory bowel diseases: CD95/Fas and beyond. Inflamm Bowel Dis. 2010;16(6):1071–6.
Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138(6):2059–72.
Whiteside TL. Tricks tumours use to escape from immune control. Oral Oncol. 2009;45(10):e199–23.
Baier PK, Wolff-Vorbeck G, Eggstein S, et al. Cytokine expression in colon carcinoma. Anticancer Res. 2005;25(3B):2135–9.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation and cancer. Cell. 2010;140(6):883–99.
Grivennikof SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70(Suppl 1):i104–8.
Lavelle EC, Murphy C, O’Neill LA, et al. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 2010;2(1):17–28.
Shao L, Kamalu O, Mayer L. Non-classical MHC class I molecules on intestinal epithelial cells: mediators of mucosal crosstalk. Immunol Rev. 2005;206:160–76.
Wells JM, Rossi O, Meijerink M, et al. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4607–14.
Frolova L, Drastich P, Rossmann P, et al. Expression of Toll-like receptor 2 (TLR2), TLR3, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem. 2008;56(3):267–74.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;1118(2):229–41.
He B, Xu W, Santini PA, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26(6):812–26.
Hershberg RM, Framson PE, Cho DH, et al. Intestinal epithelial cells use two distinct pathways for HLA class II antigen processing. J Clin Invest. 1997;100(1):204–15.
Campbell N, Yio XY, So LP, et al. The intestinal epithelial cell: processing and presentation of antigen to the mucosal immune system. Immunol Rev. 1999;172:315–24.
Colgan SP, Hershberg RM, Furuta GT, et al. Ligation of intestinal epithelial CD1d induces bioactive IL-10: critical role of the cytoplasmic tail in autocrine signalling. Proc Natl Acad Sci USA. 1999;96(24):13938–43.
Kühn R, Löhler J, Rennik D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74.
Shan M, Gentile M, Yeiser JR, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science. 2013;342(6157):447–53.
Rimoldi M, Chieppa M, Salucci V, et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol. 2005;6(5):507–14.
Iliev ID, Mileti E, Matteoli G, et al. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2009;2(4):340–50.
Chen X, Song CH, Feng BS, et al. Intestinal epithelial cell-derived integrin αβ6 plays an important role in the induction of regulatory T cells and inhibits an antigen-specific Th2 response. J Leukoc Biol. 2011;90(4):751–9.
Deng ZB, Zhuang X, Ju S, et al. Exosome-like nanoparticles from intestinal mucosal cells carry prostaglandin E2 and suppress activation of liver NKT cells. J Immunol. 2013;190(7):2579–89.
Cario E, Rosenberg IM, Brandwein SL, et al. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol. 2000;164(2):966–72.
Stadnyk AW. Intestinal epithelial cells as a source of inflammatory cytokines and chemokines. Can J Gastroneterol. 2002;16(4):241–6.
Jung HC, Eckmann L, Yang SK, et al. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95(1):55–65.
Fraser-Pitt DJ, Cameron P, McNeilly TN, et al. Phosphorylation of the epidermal growth factor receptor (EGFR) is essential for interleukin-8 release from intestinal epithelial cells in response to challenge with Escherichia coli O157: H7 flagellin. Microbiology. 2011;157(Pt 8):2339–47.
Rimoldi M, Chieppa M, Vulcano M, et al. Intestinal epithelial cells control dendritic cell function. Ann N Y Acad Sci. 2004;1029:66–74.
Bhowmick S, Chatterjee D, Chaudhuri K. Human epithelial cells stimulated with Vibrio cholerae produce shymic stromal lymphopoietin and promote dendritic cell-mediated inflammatory Th2 response. Int J Biochem Cell Biol. 2012;44(11):1779–90.
Kido M, Tanaka J, Aoki N, et al. Helicobacter pylori promotes the production of thymic stromal lymphopoietin by gastric epithelial cells and induces dendritic cell-mediated inflammatory Th2 responses. Infect Immun. 2010;78(1):108–14.
Tjabringa GS, Ninaber DK, Drijfhout JW, et al. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch Allergy Immunol. 2006;140:103–12.
Hase K, Eckmann L, Leopard JD, et al. Cell differentiation is a key determinant of cahtelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun. 2002;70(2):953–63.
Scott MG, Davidson DJ, Gold MR, et al. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune response. J Immunol. 2002;169(7):3883–91.
Wehkamp J, Schauber J, Stange EF. Defensins and cathelicidins in gastrointestinal infections. Curr Opin Gastroenterol. 2007;23(1):32–8.
Schauber J, Svanholm C, Termén S, et al. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut. 2003;52(5):735–41.
Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5(4):354–66.
Nair MG, Guild KJ, Du Y, et al. Goblet cell-derived resistin-like molecule beta augments CD4+ T cell production of IFN-gamma and infection-induced intestinal inflammation. J Immunol. 2008;181(7):4709–15.
Line A, Slucka Z, Stengrevis A, et al. Characterization of tumour-associated antigens in colon cancer. Cancer Immunol Immunother. 2002;51(10):574–82.
Chan CC, Fan CW, Kuo YB, et al. Multiple serological biomarkers for colorectal cancer detection. In J Cancer. 2010;126(7):1683–90.
Baier PK, Wimmenauer S, Hirsch T, et al. Analysis of the T-cell receptor variability of tumour-infiltrating lymphocytes in colorectal carcinomas. Tumour Biol. 1998;19(3):205–12.
Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumours predict clinical outcome. Science. 2006;313(5795):1960–4.
Ahmad M, Rees RC, Ali SA. Escape from immunotherapy: possible mechanisms that influence tumour regression/progression. Cancer Immunol Immunother. 2004;53:844–54.
Nakayama H, Kitayama J, Muto T, et al. Characterization of intracellular cytokine profile of CD4(+) T cells in peripheral blood and tumour-draining lymph nodes of patients with gastrointestinal cancer. Jpn J Clin Oncol. 2000;30(7):301–5.
Evans C, Dalgleish AG, Kumar D. Immune suppression and colorectal cancer. Aliment Pharmacol Ther. 2006;24(8):1163–77.
Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183–8.
Komima M, Morisaki T, Uchiyama A, et al. Association of enhanced cyclooxygenase-2 expression with possible local immunosuppression in human colorectal carcinomas. Ann Surg Oncol. 2001;8(5):458–65.
Gastl GA, Abrams JS, Nanus DM, et al. Interleukin-10 production by human carcinoma cell lines and its relationship to interleukin-6 expression. Int J Cancer. 1993;55:96–101.
Kucharzik T, Lugering N, Winde G, et al. Colon carcinoma cell lines stimulate monocytes and lamina propria mononuclear cells to produce IL-10. Clin Exp Immunol. 1997;110(2):296–302.
Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumours inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103.
Li Q, Pan PY, Gu P, et al. Role of immature myeloid Gr-1+ cells in the development of antitumour immunity. Cancer Res. 2004;64(3):1130–9.
Peduto Eberl L, Guillou L, Saraga E, et al. Fas and Fas ligand expression in tumour cells and in vascular smooth-muscle cells of colonic and renal carcinomas. Int J Cancer. 1999;81(5):772–8.
O’Connell J, Bennett MW, Nally K, et al. Altered mechanisms of apoptosis in colon cancer: Fas resistance and counterattack in the tumour-immune conflict. Ann N Y Acad Sci. 2000;910:178–92.
Favre-Felix N, Fromentin A, Hammann A, et al. Cutting edge: the tumour counterattack hypothesis revisited: colon cancer cells do not induce T cell apoptosis via the Fas (CD95, APO-1) pathway. J Immunol. 2000;164:5023–7.
Reinecker H, Podolsky D. Human intestinal epithelial cells express functional cytokine receptors sharing the common c chain of the interleukin 2 receptor. Proc Natl Acad Sci USA. 1995;92(18):8353–7.
Kumar N, Mishra J, Narang VS, et al. Janus kinase 3 regulates interleukin 2-induced mucosal wound repair through tyrosine phosphorylation of villin. J Biol Chem. 2007;282(42):30341–5.
Mishra J, Waters CM, Kumar N. Molecular mechanism of interleukin-2-induced mucosal homeostasis. Am J Physiol Cell Physiol. 2012;302(5):C735–47.
Kinnebrew MA, Buffie CG, Diehl GE, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36(2):276–87.
Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14(3):282–9.
Pickert G, Neufert C, Leppkes M, et al. Stat3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206(7):1465–72.
Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181(6):3733–9.
Yuan L, Wen K, Azevedo MS, et al. Virus-specific intestinal IFN-gamma producing T cell response induced by human rotavirus infection and vaccines are correlated with protection against rotavirus diarrhoea in gnotobiotic pigs. Vaccine. 2008;26(26):3322–31.
John B, Rajaqopal D, Pashine A, et al. Role of IL-12-independent and IL-12-dependent pathways in regulating generation of the IFN-gamma component of T cell responses to Salmonella typhimurium. J Immunol. 2002;169(5):2545–52.
Roberts-Thomson IC, Fon J, Uylaki W, et al. Cells, cytokines and inflammatory bowel disease: a clinical perspective. Expert Rev Gastroenterol Hepatol. 2011;5(6):703–16.
Strober W. Interactions between epithelial cells and immune cells in the intestine. Ann N Y Acad Sci. 1998;859:37–45.
Denning TL, Campbell NA, Song F, et al. Expression of IL-10 receptors on epithelial cells from murine small and large intestine. Int Immunol. 2000;12(2):133–9.
Songhet P, Barthel M, Stecher B, et al. Stromal IFN-γR-signaling modulates goblet cell function during Salmonella Typhimurium infection. PLoS ONE. 2011;6(7):e22459.
Raetz M, Hwang SH, Wilhelm CL, et al. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-γ dependent elimination of Paneth cells. Nat Immunol. 2013;14(2):136–42.
Villeret B, Brault L, Couturier-Maillard A, et al. Blockade of IL1R signalling diminishes Paneth cell depletion and Toxoplasma gondii induced ileitis in mice. Am J Clin Exp Immunol. 2013;2(1):107–16.
Zolotarevsky Y, Hecht G, Koutsouris A, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–72.
Fischer A, Gluth M, Pape UF, et al. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signalling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;304(11):G970–9.
Neurath MF, Travis SP. Mucosal healing in inflammatory bowel disease: a systemic review. Gut. 2012;61(11):1619–35.
Wittkopf N, Günther C, Martini E, et al. Cellular FLICE-like inhibitory protein secures intestinal epithelial cell survival and immune homeostasis by regulating caspase-8. Gastroenterology. 2013;. doi:10.1053/j.gastro.2013.08.059.
Günther C, Martini E, Wittkopf N, et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477(7364):335–9.
Nenci A, Becker C, Wullaert A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446(7135):557–61.
Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–38.
Gillen CD, Walmsley RS, Prior P, et al. Ulcerative colitis and Crohn’s disease: a comparison of colorectal cancer risk in extensive colitis. Gut. 1994;35(11):1590–2.
Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–34.
Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–25.
Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumourigenesis. Cancer Cell. 2009;15(2):91–102.
Lee H, Herrmann A, Deng JH, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumours. Cancer Cell. 2009;15:283–93.
Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–71.
Balwill F, Joffroy C. TNF: a tumour-suppressing factor or a tumour-promoting factor? Future Oncol. 2010;6(12):1833–6.
Carswell EA, Old LJ, Kassel RL, et al. An endotoxin-induced serum factor that causes necrosis of tumours. Proc Natl Acad Sci USA. 1975;72(9):3666–70.
Balkwill FR, Lee A, Aldam G, et al. Human tumour xenografts treated with recombinant human tumour necrosis factor alone or in combination with interferons. Cancer Res. 1986;46(8):3990–3.
Brouckaert PG, Leroux-Roels CG, Guisez Y, et al. In vivo anti-tumour activity of recombinant human and murine TNF, alone and in combination with murine IFN-gamma, on a syngeneic murine melanoma. Int J Cancer. 1986;38(5):763–9.
Selby P, Hobbs S, Viner C, et al. Tumour necrosis factor in man: clinical and biological observations. Br J Cancer. 1987;56(6):803–8.
Blick M, Sherwin SA, Rosenblum M, et al. Phase I study of recombinant tumor necrosis factor in cancer patients. Cancer Res. 1987;47(11):2986–9.
Lienard D, Ewalenko P, Delmotte JJ, et al. High-dose recombinant tumor necrosis factor alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanoma and sarcoma. J Clin Oncol. 1992;10(1):52–60.
Sugarman BJ, Aggarwal BB, Hass PE, et al. Recombinant human tumour necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science. 1985;230(4728):943–5.
Sakai H, Yamada Y, Shimizu M, et al. Genetic ablation of Tnfalpha demonstrates no detectable suppressive effect on inflammation-related mouse colon tumorigenesis. Chem Biol Interact. 2010;184(3):423–30.
Rivas MA, Carnevale RP, Proietti CJ, et al. TNF alpha acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp Cell Res. 2008;314(3):509–29.
Liu RY, Fan C, Liu G, et al. Activation of p38 mitogen-activated protein kinase is required for tumor necrosis factor-alpha -supported proliferation of leukemia and lymphoma cell lines. J Biol Chem. 2000;275(28):21086–93.
Popivanova BK, Kitamura K, Wu Y, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. N J Clin Invest. 2008;118(2):560–70.
Min HY, Chung HJ, Kim EH, et al. Inhibition of cell growth and potentiation of tumour necrosis factor-α (TNF-α)-induced apoptosis by a phenanthroindolizidine alkaloid antofine in human colon cancer cells. Biochem Pharmacol. 2012;80(9):1356–64.
Park ES, Yoo JM, Yoon DY, et al. IL-32γ enhances TNF-α-induced cell death in colon cancer. Mol Carcinog. 2012;. doi:10.1002/mc.21990.
Ogawa S, Nagoa M, Kanehiro H, et al. The breakdown of apoptotic mechanism in the development and progression of colorectal carcinoma. Anticancer Res. 2004;24:1569–79.
Ryu BK, Lee MG, Chi SG, et al. Increased expression of cFLIP(L) in colonic adenocarcinoma. J Pathol. 2001;194:15–9.
Acknowledgment
This work was supported by the Interdisciplinary Center for Clinical Research Erlangen, project D19, the European Community’s 7th Framework Program (acronym BTCure) and the DFG (SFB796, SPP1656, clinical research unit KFO257, BE3686/2).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wittkopf, N., Neurath, M.F. & Becker, C. Immune-epithelial crosstalk at the intestinal surface. J Gastroenterol 49, 375–387 (2014). https://doi.org/10.1007/s00535-013-0929-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-013-0929-4