
Abstract
Biosimilars are biological medicines that have been shown to be similar to a reference biological medicine that has already been approved for use. Development of biosimilars is based on a “totality of evidence” approach that involves a series of steps by which biosimilars must demonstrate similarity to a reference product in all aspects of the drug and eliminate any remaining uncertainties. Clinical studies are then considered confirmatory and are performed to show that there are no clinically meaningful differences compared with the reference product in a sensitive patient population. The recombinant human granulocyte colony-stimulating factor (G-CSF) biosimilar EP2006/Zarxio® (filgrastim-sdnz) became the first FDA-approved biosimilar in 2015. This review evaluates how clinical equivalence can be demonstrated with G-CSF biosimilars through the identification of “sensitive” study populations and endpoints. Patients with non-metastatic breast cancer treated in the (neo)adjuvant setting represent a potentially homogenous population, making this a suitable sensitive indication for assessing filgrastim and pegfilgrastim biosimilars compared with reference products. This review includes clinical trials of G-CSF biosimilars in breast cancer, focusing on key aspects of the trials that were necessary to accurately demonstrate clinical equivalence and enable extrapolation to relevant indications, based on guidelines and biostatistical principles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
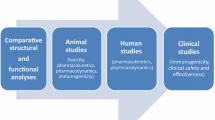
Biosimilars are biological medicines that have been shown to be similar to a reference biological medicine that has already been approved for use [1]. As patents for biological drugs expire, there are increased opportunities for the development of biosimilars and, as such, biosimilars are becoming increasingly available, particularly in oncology [2]. The development of biosimilars is significantly more complex than the development of small molecule generic drugs [2], but the principles for their development and approval, based on a “totality of evidence” approach, are now well established. Totality of evidence involves a series of steps by which biosimilars must demonstrate similarity to a reference product in all aspects of the drug and eliminate any remaining uncertainties [3]. This sequential process must include comparative structural and functional characterization, nonclinical evaluation, and clinical studies to compare human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical safety and immunogenicity data, and typically comparative clinical efficacy studies [1, 4]. This stepwise approach is essential because clinical studies are generally the least sensitive means to detect differences between two biological products. Clinical studies are nonetheless vital to confirm that there are no clinically meaningful differences in terms of biological activity, safety, and immunogenicity compared with the reference product [5].
In Europe, 12 biosimilars have been approved in oncology indications since the approval of the first biosimilar in this field, Binocrit® (epoetin alfa), in 2007 [6]. Although biosimilars have been slower to enter the US market, the recombinant human granulocyte colony-stimulating factor (G-CSF) biosimilar EP2006/Zarxio® (filgrastim-sdnz) became the first FDA-approved biosimilar in 2015 [7]. This review aims to evaluate how clinical equivalence can be demonstrated with G-CSF biosimilars through the identification of “sensitive” study populations and endpoints, and to consider how this enables subsequent extrapolation to other relevant indications. We reviewed clinical trials of G-CSF biosimilars in breast cancer, focusing on key aspects of the trials that were necessary to accurately demonstrate clinical equivalence and enable extrapolation to relevant indications, based on guidelines and biostatistical principles.
Extrapolation
The European Medicines Agency (EMA) defines extrapolation as “extending information and conclusions available from studies in one or more subgroups of the patient population (source population) … to make inferences for another subgroup of the population (target population), or condition or product, thus reducing the need to generate additional information… to reach conclusions for the target population” [8]. Extrapolation must be scientifically justified in order to support a determination of biosimilarity for each additional indication; this is dependent on multiple factors that must be consistent across each indication, including similarity in structural and functional properties, the knowledge of the mechanism of action, similar PK and bio-distribution, immunogenicity, and expected toxicities [4].
An important consideration regarding extrapolation is that it is already an established scientific principle in drug regulation, for example, in a case of a major change of a manufacturing process, or when data from intravenous formulations are extrapolated to a new subcutaneous formulation [3]. Furthermore, extrapolation is also well accepted when a drug has been evaluated in a randomized clinical trial setting with strict inclusion and exclusion criteria, and then is extrapolated to real-world patients outside the restrictions of a clinical trial.
Guidelines for biosimilar development stipulate that in order to permit extrapolation, the clinical data collected using the totality of evidence approach must be in a “sensitive indication” [1, 2, 4] (Table 1). A sensitive indication is a patient population in which the treatment being assessed has a large effect on the relevant endpoint so that a difference between biosimilar and reference product will most easily be detected. Likewise, an immunocompetent study population is required to allow the meaningful evaluation of immunogenicity [1, 4].
Epoetin is a good example of selecting a sensitive indication in biosimilar development, which enables extrapolation to other indications. Binocrit® (epoetin alfa biosimilar) has been approved in Europe since 2007 [6] for the treatment of chemotherapy-induced anemia and renal anemia [9]. Patients with renal anemia without any major complications or comorbidities that may alter the response to epoetin offer a sensitive population to assess biosimilarity since potential differences in efficacy between the reference and biosimilar may be more easily shown in this population rather than in cancer patients undergoing chemotherapy who may be immunosuppressed and have variable responses to epoetin [10]. Furthermore, this indication was particularly relevant since renal anemia patients are the population at risk of developing pure red cell aplasia (PRCA) with epoetin treatment and no cases of PRCA have been reported in oncology patients receiving epoetin [3]. Extrapolation of the use of epoetin from renal anemia to cancer patients was scientifically justified since the biological effect is mediated by the same mechanism of action. The totality of evidence approach established that biosimilar epoetin alfa is similar to the reference medicine and, as such, extrapolation of renal anemia to chemotherapy-induced anemia was permitted [9].
Rationale for (neo)adjuvant breast cancer as a sensitive indication for assessment of G-CSF biosimilarity
G-CSF has numerous indications, including reduction in neutropenia/febrile neutropenia in patients receiving cytotoxic chemotherapy; mobilization of peripheral blood progenitor cells; treatment of severe congenital, cyclic, or idiopathic neutropenia; and treatment of persistent neutropenia in HIV patients [11,12,13,14,15,16]. However, G-CSF acts via the same mechanism of action across all associated patient populations, through selective binding of the G-CSF receptor [5, 17]. Therefore, if a study directly compares reference and biosimilar G-CSF in a sensitive population and demonstrates similarity, this supports extrapolation across all indications as part of the totality of evidence concept [3, 18].
Selection of a sensitive population in which to investigate potential differences between a reference medicine and a proposed biosimilar includes identification of a homogeneous population. Homogeneous populations allow any difference in response between reference and biosimilar to be attributed to product characteristics and reduce the likelihood that it is due to individual variation [19]. Increased homogeneity within a population contributes to increased sensitivity, allowing more accurate assessment of similarity compared with heterogeneous populations.
One of the indications for G-CSF is to decrease the risk of febrile neutropenia in patients with non-myeloid malignancies undergoing chemotherapy. Within this indication, patients receiving (neo)adjuvant treatment for breast cancer can be considered a sensitive cohort in which to assess biosimilar compared with reference G-CSF since it provides a homogenous patient population [18,19,20]. A key feature of this homogeneity is that unlike patients with metastatic breast cancer, patients with (neo)adjuvant disease have not received prior chemotherapy. This means that they exhibit less inter-patient variation in terms of potential for treatment-related toxicity and other confounding factors such as disease burden, location of metastases, and phenotype of metastatic cells [21]. This also means that patients with (neo)adjuvant breast cancer are, in general, representative of breast cancer patients worldwide, provided disease and treatment characteristics are similar [18,19,20,21]. Furthermore, these patients have not yet received treatment that likely differs from region to region. In addition, unlike previously treated patients, (neo)adjuvant patients have not experienced previous chemotherapy-induced immunosuppression and, as such, are a more sensitive population in which to assess risk of immunogenicity.
TAC (docetaxel, doxorubicin, and cyclophosphamide) chemotherapy is recommended in international treatment guidelines as one of the standard (neo)adjuvant chemotherapy regimens for patients with breast cancer due to its documented efficacy [22]. TAC has a proven dose-limiting hematological toxicity with grade 3–4 neutropenia in 65.5% patients [23], a median duration of severe (grade 4) neutropenia (DSN) of 7 days [24], and febrile neutropenia reported in 24–34% of patients [23,24,25,26] without G-CSF support. Treatment guidelines require primary prophylaxis with G-CSF as supportive care for TAC chemotherapy [27,28,29] with a proven substantial effect in this setting, reducing mean DSN to 1.4 days (95% confidence interval [CI] 1.1, 1.7) [30].
Demonstrating clinical equivalence of G-CSF during randomized controlled trials in (neo)adjuvant breast cancer
Endpoints measured are a key consideration when planning confirmatory clinical studies comparing biosimilar and reference medicines. Sensitive endpoints should assess biological activity of the proposed biosimilar, as opposed to treatment outcomes, to allow similarity to be assessed more accurately [1]. DSN can be considered a sensitive endpoint in assessing biosimilarity of G-CSF in (neo)adjuvant breast cancer. Due to its dependence on G-CSF efficacy, any variation in DSN between homogeneous treatment groups can be considered to be a direct consequence of differences between activity of reference and biosimilar rhG-CSF. This sensitivity compared with other endpoints (e.g., infections, febrile neutropenia) can also be attributed to its continuous nature and frequent repeat sampling. Furthermore, risk of infection is directly proportional to severity and duration of neutropenia [31], making DSN a clinically relevant endpoint.
Clinical studies designed to assess potential differences between a reference medicine and a proposed biosimilar are typically designed to show equivalence of the two treatments. Equivalence in this sense means that the efficacies of the two products under assessment are similar to the extent that neither could be considered superior or inferior to the other [32]. In equivalence trials, the objective is to demonstrate that the biosimilar (b) is not meaningfully different to the reference (r), in terms of an endpoint (μ):
where Δ represents the equivalence margin, defined as “the maximum tolerable difference considered to be clinically acceptable” [32].
When assessing biosimilarity, it is essential that a clinically relevant and meaningful equivalence margin is established, i.e., the range over which the efficacies can be considered equivalent [32]. Identification of an appropriate equivalence margin is dependent on the specific characteristics of the reference product and its therapeutic class [19]. Therapeutic equivalence is concluded if the 95% confidence interval is completely contained within the equivalence margin. This is statistically equivalent to calculating two independent one-sided tests at a 2.5% alpha level (one in each direction), of which both have to be successful [33].
A second key consideration when assessing biosimilarity is ensuring that the trial is sufficiently powered to avoid making a type II error, i.e., incorrectly claiming that there is no difference between two treatment groups. This is dependent on factors including level of type I error (typically p = 0.05), level of type II error (p = 0.10 or 0.20), standard deviation (estimated from published or preliminary data), an estimation of the true value of μ r − μ b, and the equivalence margin [33]. Based on these biostatistical considerations, calculations were performed to identify an appropriate equivalence margin and sample size necessary to assess clinical equivalence in DSN between reference and biosimilar G-CSF in patients with (neo)adjuvant breast cancer (Table 2). Using these calculations, it can be established that at a significance level of 0.05%, a power of 90%, an equivalence limit of 0.5 days difference in DSN, and a standard deviation of 1.5, 86 patients are required per treatment group in order to robustly assess equivalence.
Patients with breast cancer represent a sensitive population for clinically evaluating all G-CSF medicines, including biosimilars. In accordance with these considerations, multiple randomized controlled trials (RCTs) have been conducted to demonstrate equivalence between biosimilar and reference G-CSF in breast cancer (Table 3). Clinical studies were also performed to compare reference pegfilgrastim with reference filgrastim in patients with breast cancer [34,35,36]. Sensitive endpoints examined include DSN, incidence of/hospitalization due to febrile neutropenia; incidence of infections; depth and time of absolute neutrophil count (ANC) nadir; and time to ANC recovery [5, 18].
Based on the totality of evidence provided, and the use of sensitive patient populations, studies demonstrating clinical equivalence of biosimilar G-CSF in breast cancer can be extrapolated to support clinical equivalence with reference filgrastim in other tumor types and indications. Given the availability for clinically relevant PD parameters for G-CSF treatment (ANC, CD34+ cell count), highly sensitive PK/PD studies can waive the need for a comparative phase III trial for regulatory approval including full extrapolation in Europe under certain circumstances. For example, following demonstration of comparability in structural and functional attributes and in PK/PD characteristics compared with reference filgrastim in healthy volunteers, with a confirmatory safety single-arm phase III trial in patients with breast cancer, the biosimilar filgrastim Zarzio® was approved by the EMA for the same indications as reference biosimilar [13, 16]. In the USA, the FDA requested an additional randomized controlled clinical trial. Therefore, and following a further head-to-head comparator study in patients with breast cancer vs reference filgrastim [18], Zarzio® (marketed as Zarxio® in the USA) was subsequently approved for the same indications by the FDA [5].
This approach, taken to confirm the equivalence of reference and biosimilar G-CSF in a sensitive population, is now being used to show equivalence between biosimilar and reference pegfilgrastim. To date, two confirmatory phase III trials, PROTECT-1 and PROTECT-2, have provided evidence of therapeutic equivalence according to the abovementioned sensitive endpoints in a total of 622 patients with (neo)adjuvant breast cancer [40, 41]. However, regulatory authorities have determined that further trials are necessary to address unanswered questions, such as a potential lack of equivalence in the concentrations of pegfilgrastim compared with biosimilar pegfilgrastim in blood [44].
Conclusions
Using the rigorous “totality of evidence” approach, clinical equivalence between reference and biosimilar products can be established in a single sensitive population and reliably extrapolated to further indications. (Neo)adjuvant, non-metastatic breast cancer is a suitable sensitive patient population for assessing filgrastim and pegfilgrastim biosimilars compared with reference products.
References
European Medicines Agency. Guideline on similar biological medicinal products. EMA, London, October 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Last Accessed June 2017
Tabernero J, Vyas M, Giuliani R et al (2016) Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open 1:e000142. https://doi.org/10.1136/esmoopen-2016-000142
Weise M, Kurki P, Wolff-Holz E et al (2014) Biosimilars: the science of extrapolation. Blood 124(22):3191–3196
FDA Biosimilars Guidance 2015. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for Industry. Available from: https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm290967.htm Last Accessed 2nd May 2017
Holzmann J, Balser S, Windisch J (2016) Totality of the evidence at work: the first U.S. biosimilar. Expert Opin Biol Ther 16(2):137–142
GaBI biosimilars approved in Europe. Last updated March 2017. Available from http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe Last Accessed 4th May 2017
FDA Oncologic Drugs Advisory Committee Meeting. ZARXIO® (filgrastim). January 7, 2015
European Medicines Agency. Concept paper on extrapolation of efficacy and safety in medicine development. EMA, London, March 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/04/WC500142358.pdf Last Accessed June 2017
Binocrit® EPAR. Last updated May 2016. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000725/human_med_000675.jsp&mid=WC0b01ac058001d124 Last Accessed 11th May 2017
European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins (revision). EMA, London, March 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089474.pdf Last Accessed June 2017
Accofil EPAR. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003956/human_med_001798.jsp&mid=WC0b01ac058001d124 Last Accessed 11th May 2017
Grastofil EPAR. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002150/human_med_001688.jsp&mid=WC0b01ac058001d124 Last Accessed 11th May 2017
Neupogen® Summary of product characteristics. October 2015. Available from: https://www.medicines.org.uk/emc/medicine/27485 Last Accessed 3rd May 2017
Nivestim EPAR. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001142/human_med_001344.jsp&mid=WC0b01ac058001d124 Last Accessed 11th May 2017
Ratiograstim EPAR. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000825/human_med_001015.jsp&mid=WC0b01ac058001d124 Last Accessed 11th May 2017
Zarzio® EPAR. Last updated 06/02/2017. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000917/human_med_001170.jsp&mid=WC0b01ac058001d124 Last Accessed 3rd May 2017
Sörgel F, Schwebig A, Holzmann J et al (2015) Comparability of biosimilar filgrastim with originator filgrastim: protein characterization, pharmacodynamics, and pharmacokinetics. BioDrugs 29(2):123–131
Blackwell K, Semiglazov V, Krasnozhon D et al (2015) Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol 26(9):1948–1953
Jackisch C, Scappaticci FA, Heinzmann D et al (2015) Neoadjuvant breast cancer treatment as a sensitive setting for trastuzumab biosimilar development and extrapolation. Future Oncol 11(1):61–71
Socinski MA, Curigliano G, Jacobs I et al (2015) Clinical considerations for the development of biosimilars in oncology. MAbs 7(2):286–293
Cortés J, Curigliano G, Diéras V (2014) Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat 144(2):233–239
Senkus E, Kyriakides S, Ohno S et al (2015) Primary breast cancer: ESCO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals Oncol 26(Suppl. 5):v8–v30
Martin M, Pienkowski T, Mackey J et al (2005) Adjuvant docetaxel for node-positive breast cancer. N Engl J Med 352(22):2302–2313
Nabholtz JM, Mackey JR, Smylie M et al (2001) Phase II study of docetaxel, doxorubicin, and cyclophosphamide as first-line chemotherapy for metastatic breast cancer. J Clin Oncol 19:314–321
Martín M, Lluch A, Seguí MA et al (2006) Toxicity and health-related quality of life in breast cancer patients receiving adjuvant docetaxel, doxorubicin, cyclophosphamide (TAC) or 5-fluorouracil, doxorubicin and cyclophosphamide (FAC): impact of adding primary prophylactic granulocyte-colony stimulating factor to the TAC regimen. Ann Oncol 17(8):1205–1212
Smith TJ, Khatcheressian J, Lyman GH et al (2006) 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 24(19):3187–3205
Aapro M et al (2011) 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Clin Oncol 47:8–32
National Comprehensive Cancer Network (NCCN) 2016. Clinical practice guidelines in oncology. Myeloid growth factors. Version 1.2016
Smith TJ, Bohlke K, Lyman GH et al (2015) Recommendations for the use of WBC growth factors: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol 33:3199–3212
Burris HA, Belani CP, Kaufman PA et al (2010) Pegfilgrastim on the same day versus next day of chemotherapy in patients with breast cancer, non-small-cell lung cancer, ovarian cancer, and non-Hodgkin’s lymphoma: results of four multicenter, double-blind, randomized phase II studies. J Oncol Pract 6(3):133–140
Bodey GP (1984) Antibiotics in patients with neutropenia. Arch Intern Med 144(9):1845–1851
Walker E, Nowacki AS (2011) Understanding equivalence and noninferiority testing. J Gen Intern Med 26(2):192–196
Altman DG (1991) Practical statistics for medical research, Chapman & Hall/CRC, London. ISBN 9780412276309
Green MD, Koelbl H, Baselga J et al (2003) A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 14(1):29–35
Holmes FA, O'Shaughnessy JA, Vukelja S et al (2002a) Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high-risk stage II or stage III/IV breast cancer. J Clin Oncol 20(3):727–731
Holmes FA, Jones SE, O'Shaughnessy J et al (2002b) Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 13(6):903–909
del Giglio A, Eniu A, Ganea-Motan D et al (2008) XM02 is superior to placebo and equivalent to Neupogen in reducing the duration of severe neutropenia and the incidence of febrile neutropenia in cycle 1 in breast cancer patients receiving docetaxel/doxorubicin chemotherapy. BMC Cancer 8:332
Waller CF, Semiglazov VF, Tjulandin S et al (2010) A phase III randomized equivalence study of biosimilar filgrastim versus Amgen filgrastim in patients receiving myelosuppressive chemotherapy for breast cancer. Onkologie 33(10):504–511
Buchner A, Elsässer R, Bias P (2014) A randomized, double-blind, active control, multicenter, dose-finding study of lipegfilgrastim (XM22) in breast cancer patients receiving myelosuppressive therapy. Breast Cancer Res Treat 148(1):107–116
Blackwell K, Donskih R, Jones CM et al (2016) A comparison of proposed biosimilar LA-EP2006 and reference pegfilgrastim for the prevention of neutropenia in patients with early-stage breast cancer receiving myelosuppressive adjuvant or neoadjuvant chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21(7):789–794
Harbeck N, Lipatov O, Frolova M et al (2016) Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12(11):1359–1367
Hegg R, Mattar A, Matos-Neto JN et al (2016) A phase III, randomized, non-inferiority study comparing the efficacy and safety of biosimilar filgrastim versus originator filgrastim for chemotherapy-induced neutropenia in breast cancer patients. Clinics (Sao Paulo) 71(10):586–592
Waller CF, Blakeley C, Pennella E et al (2016) Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-Neulasta® in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol 27(suppl_6):1433O
European Medicines Agency. Withdrawal of the marketing authorisation application for Zioxtenzo (pegfilgrastim). EMA, London, January 2017. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2017/01/WC500220231.pdf Last Accessed June 2017
Acknowledgements
Editorial support was provided by Caroline McGown of the Spirit Medical Communications Ltd., supported by the Sandoz GmbH, Kundl, Austria. Final approval of the manuscript rested solely with the scientific authors.
Funding
This work was supported by the Sandoz GmbH, Kundl, Austria. Authors received no monetary compensation in return for their work on this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Matti Aapro: Amgen (honoraria, speakers bureau, expert testimony), Helsinn Healthcare (advisory role, speakers bureau, research funding), Hospira (advisory role, speakers bureau, research funding), Teva (advisory role, speakers bureau), Merck KGaA (advisory role), Merck (advisory role), Sandoz (advisory role, speakers bureau, research funding), Pierre Fabre Medicament (advisory role, speakers bureau, research funding), Vifor Pharma (advisory role, speakers bureau), Tesaro (advisory role, speakers bureau), Novartis (speakers bureau, research funding), Roche (speakers bureau), Johnson & Johnson (speakers bureau)
Andriy Krendyukov: Hexal AG (employment)
Martin Schiestl: Hexal AG (employment)
Nadia Hoebel: Hexal AG (employment)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Krendyukov, A., Schiestl, M., Höbel, N. et al. Clinical equivalence with G-CSF biosimilars: methodologic approach in a (neo)adjuvant setting in non-metastatic breast cancer. Support Care Cancer 26, 33–40 (2018). https://doi.org/10.1007/s00520-017-3861-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-017-3861-y