
Abstract
Plants and algae have a complex life history that transitions between distinct life forms called the sporophyte and the gametophyte. This phenomenon—called the alternation of generations—has fascinated botanists and phycologists for over 170 years. Despite the mesmerizing array of life histories described in plants and algae, we are only now beginning to learn about the molecular mechanisms controlling them and how they evolved. Epigenetic silencing plays an essential role in regulating gene expression during multicellular development in eukaryotes, raising questions about its impact on the life history strategy of plants and algae. Here, we trace the origin and function of epigenetic mechanisms across the plant kingdom, from unicellular green algae through to angiosperms, and attempt to reconstruct the evolutionary steps that influenced life history transitions during plant evolution. Central to this evolutionary scenario is the adaption of epigenetic silencing from a mechanism of genome defense to the repression and control of alternating generations. We extend our discussion beyond the green lineage and highlight the peculiar case of the brown algae. Unlike their unicellular diatom relatives, brown algae lack epigenetic silencing pathways common to animals and plants yet display complex life histories, hinting at the emergence of novel life history controls during stramenopile evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The alternation of generations
The alternation of generations is a developmental phenomenon where two distinct life forms—the sporophyte and gametophyte—alternate in the life cycle of plants and algae. The sporophyte is diploid and gives rise to haploid spores at meiosis that go on to develop into gamete-producing gametophytes. The union of gametes at fertilization initiates the formation of the diploid sporophyte to complete this so-called haplo-diplontic life cycle (Fig. 1). The classical studies of Wilhelm Hofmeister were the first to establish the universal occurrence of two life forms in plants (Hofmeister 1851, 1862). Hofmeister keenly recognized the presence of two separate, free-living generations in the land plant life cycle, despite substantial varying morphologies among the major plant groups. What ensued was a debate among scientists of the late nineteenth century about the origin of these alternating generations, and the key discovery that these alternations also included changes in chromosome number or ploidy. The history and outcome of these classical debates can be delved into by the reader in excellent in-depth reviews on the subject (Bell 1989; Haig 2008).

Life cycle schemes in living organisms. Haplo-diplontic life cycles are characterized by mitotic divisions in both the diploid zygote and haploid meiotic spores, which produce distinct and sometimes free-living sporophytes and gametophytes, respectively. The diploid zygote immediately undergoes meiosis in haplontic life cycles, where mitotic divisions of the resulting haploid spores generate a large multicellular individual or more unicellular haploid cells. Conversely, the diploid zygote in organisms with a diplontic life cycle divides mitotically to produce a multicellular individual or more unicellular diploid cells. Once diploid cells of diplontic organisms undergo meiosis, the resulting haploid cells directly differentiate into the gametes
An obvious advantage of a complex life cycle is the adaptation of life forms to distinct functional roles (Dickinson and Grant-Downton 2009). The sporophyte and gametophyte often exhibit substantial differences in physical size, cell types and duration spent in either phase (Bell 1989). A striking evolutionary trend observed in the plant and brown algal lineages, both of which evolved complex multicellularity, is a tendency for a dominant diploid phase and a concomitant reduction in the number of mitotic divisions and cell types of the haploid phase (Rudall 2006a; Rudall and Bateman 2007a). The life cycle of angiosperms serves as an apt example, where the diploid sporophyte represents the bulk of the vegetative plant body while the inconspicuous pollen grain and ovule represent the male and female gametophytes, respectively. A perspective on the adaptive qualities of alternating life cycles and the emergence of the sporophyte as the dominant phase can be garnered elsewhere (Graham and Wilcox 2000; Haig and Wilczek 2006).
As a developmental process, the alternation of generations is remarkable in that a single genome can express multiple morphologically distinct life forms. While fertilization and meiosis represent the transition checkpoint between these life forms (Fig. 1), changes in ploidy level alone do not appear to be sufficient to initiate these developmental transitions. Haploid sporophytes are readily produced through in vitro anther culture in several angiosperms (Hu and Guo 1999; Lv et al. 2020; Wang et al. 2021), while diploid gametophytes are observed in some developmental mutants in the brown algae (Coelho et al. 2011a; Arun et al. 2019). Moreover, the gametes in several algal species can develop into haploid sporophytes through parthenogenesis and thereby bypass fertilization altogether (Coelho et al. 2011b). How the marked differences in gene expression, development and morphology are established between the gametophyte and sporophyte is poorly understood and thus remains an important question in developmental biology.
Patterns of DNA and histone methylation, which control genomic activity through transcriptional repression, are reprogrammed multiple times during germline differentiation and early embryogenesis in mammals (Morgan et al. 2005). This erasure of epigenetic marks is called epigenetic reprogramming and it serves to reshape the transcriptional landscapes that specify the gametes and development of the embryo (Feng et al. 2010). It thus stands to reason that the epigenetic patterns established in the gametophyte or sporophyte undergo reprogramming during the alternation of generations. There have been extensive cellular and molecular investigations of the sporophyte and gametophyte, particularly in plants, but these tend to occur in isolation and often without consideration of the alternation of generations (Dickinson and Grant-Downton 2009). At the level of epigenetics and genomics, less progress has been made due to the difficulty of isolating the microscopic cells of the less dominant generation. Here, we undertake a comprehensive review of epigenetic studies in light of the evolution and control of the alternation of generations. We critically assess the existing literature and highlight more recent studies that have directly addressed this phenomenon in Arabidopsis, then postulate how epigenetic silencing might have been adapted to regulate life history transitions during plant evolution. We end by discussing the phylogenetically distinct stramenopile lineage and highlight the independent evolutionary origin of the alternation of generations in brown algae.
Epigenetic modifications are few and far between
There is much confusion in the literature over the term ‘epigenetic’ since it is often used to encompass all forms of DNA and histone modification (Bird 2007). The most accurate and accepted definition of epigenetics defines it as ‘the study of heritable phenotypes that do not alter the DNA sequence’ (Bonasio et al. 2010; Arimondo et al. 2019). Just as genetic information encoded by DNA is inherited from cell-to-cell, so too must an epigenetic modification be faithfully restored during each cell cycle (Reinberg and Vales 2018). Bearing this in mind, only a handful of DNA and histone modifications should be considered truly epigenetic. With this in mind, we will briefly introduce the most bona fide examples of epigenetic marks that will form the basis for our ensuing discussion.
5′-methylcytosine (5mC)—the methylated form of the nucleotide cytosine—is the most widely studied epigenetic modification (Holliday and Pugh 1975; Bird 2002). In animals, 5mC occurs at CG dinucleotides, while in plants it can occur in CG, CHG and CHH dinucleotide contexts (where H denotes A, C or T) (Kawashima and Berger 2014). Methylation in each context plays distinct biological functions and is deposited by dedicated families of DNA methyltransferases (Law and Jacobsen 2010; Stroud et al. 2014). DNA methylation is largely confined to promoters and heterochromatic regions, where it mediates gene regulation, transposable element (TE) silencing and genome stability (Zhang et al. 2018). In addition, CG methylation is also enriched over the gene body of expressed genes (Bewick and Schmitz 2017). The symmetric nature of CG methylation results in its semi-conservative segregation during DNA replication, such that it is faithfully restored on newly synthesized strands of DNA through a template-copying mechanism (Zhu and Reinberg 2011). In animals, CG methylation is re-established by DNA methyltransferase 1 (DNMT1) through its recruitment to the replication fork by PCNA and UHRF1 where it preferentially methylates hemi-methylated DNA (Bostick et al. 2007).
In plants, VIM proteins represent the orthologs of mammalian UHRF1 that probably also act to recruit MET1, the plant ortholog of mammalian DNMT1 that restores CG methylation (Woo et al. 2008), while symmetric CHG methylation is restored by Chromomethylase 3 (CMT3) (Lindroth et al. 2001). In contrast, asymmetric CHH methylation is deposited by Domains Rearranged Methyltransferase 2 (DRM2) through RNA-directed DNA methylation (RdDM) and by CMT2 (Matzke et al. 2009; Zemach et al. 2013). A feedback loop mechanism is required where CMT2 and CMT3 recognize H3K9me2 to methylate neighboring CHH and CHG sites while the H3K9 methyltransferase KRYPTONITE (KYP) recognizes methylated cytosines, further reinforcing the transcriptional silencing of heterochromatin (Wilkins and Holliday 2009; Du et al. 2014; Niklas et al. 2014; Lenormand et al. 2016). In the yeast Cryptococcus neoformans, CG methylation is propagated by the maintenance methyltransferase DNMT5 in the absence of a de novo methyltransferase and also involves an H3K9 methylation loop (Catania et al. 2020). Symmetric CG and CHG methylation thus represent mitotically heritable epigenetic marks in plants, whereas non-symmetric CHH methylation requires de novo establishment during each cell cycle.
Chromatin disassembly during DNA replication results in the recycling of parental nucleosomes onto nascent strands of chromatin (Petryk et al. 2018; Reverón-Gómez et al. 2018; Schlissel and Rine 2019). This re-partitioning of modified nucleosomes provides a means for the epigenetic inheritance and restoration of histone modifications from mother-to-daughter cells (Petryk et al. 2018; Reverón-Gómez et al. 2018; Schlissel and Rine 2019). Euchromatic histone modifications like H3K9ac and H3K27ac as well as H3K4me1, H3K4me3 and H3K36me3, all of which play crucial roles in transcription, do not undergo maintenance across the cell cycle (Henikoff and Shilatifard 2011; Pérez-Lluch et al. 2015). Only two types of histone modification—the methylated forms of H3K9 and H3K27—have been demonstrated to be transmissible through cell division (Zhu and Reinberg 2011). The mitotic heritability of H3K9 methylation has been demonstrated in fission yeast (Grewal and Klar 1996; Nakayama et al. 2001; Grewal and Jia 2007; Audergon et al. 2015; Torres-Garcia et al. 2020), which involves a self-sustaining ‘read-write’ mechanism that is mediated by the recruitment of H3K9 methyltransferases to the replication fork (Reese et al. 2003; Sarraf and Stancheva 2004; Estève et al. 2006; Loyola et al. 2009; Li et al. 2011). In plants, H3K9me2 represents the equivalent of H3K9me3 and is catalyzed by KRYPTONITE (KYP)/SU(VAR)3-9 HOMOLOG 4 (SUVH4), SUVH5 and SUVH6 (Jackson et al. 2002; Du et al. 2012, 2014, 2015). Although likely to be conserved, direct evidence for the recruitment of these H3K9 methyltransferases to the replication fork in plants, which would mediate epigenetic inheritance of H3K9me2, has yet to be demonstrated.
A similar read–write mechanism exists to propagate H3K27 methylation across cell divisions. H3K27me3 is deposited by the widely conserved Polycomb Repressive Complex 2 (PRC2) (Hennig and Derkacheva 2009). PRC2 was first discovered from genetic screens in Drosophila melanogaster as a key regulator of homeotic genes during development (Schwartz and Pirrotta 2007). PRC2 is comprised of four proteins, with H3K27me3 deposition carried out by homologs of Drosophila SET domain methyltransferase Enhancer of Zeste (E(z)) (Schwartz and Pirrotta 2007). PRC2-mediated silencing of developmental genes is highly conserved across animals and plants and is essential for maintaining cellular identity during growth and development (Margueron and Reinberg 2011). In animals, PRC2 localizes to replication foci throughout S-phase where it propagates H3K27me3 domains across the cell cycle using pre-existing H3K27me3-marked nucleosomes as a template (Hansen et al. 2008; Margueron et al. 2009; Coleman and Struhl 2017; Laprell et al. 2017).
Similarly, the loss of PRC2 from chromatin dilutes H3K27me3 in a cell cycle–dependent manner during plant cell fate determination (Sun et al. 2014). PRC2 subunits have been shown to interact with components of the replication fork in Arabidopsis, which include the E(z) ortholog CURLY LEAF (CLF) (Jiang and Berger 2017). H3K27me3 maintenance involves the initial K27 mono-methylation of replicative histone H3.1 by the plant-specific SET domain methyltransferases ARABIDOPSIS TRITHORAX-RELATED PROTEIN 5 and 6 (ATXR5/6) (Jacob et al. 2014; Jiang and Berger 2017). The coupling of read–write activity in H3K27me3 inheritance also involves LIKE HETEROCHROMATIN PROTEIN 1 (LHP1), which binds H3K27me3 and interacts with the PRC2 subunit MULTICOPY SUPRESSOR OF IRA 1 (MSI1) (Derkacheva et al. 2013), and potentially also FERTILIZATION INDEPENDENT ENDOSPERM (FIE) (Ohad et al. 1999), the plant ortholog of the EED subunit that tethers PRC2 to H3K27me3 in animals (Margueron et al. 2009). This leads to a model where ATXR5/6 and multiple Polycomb group proteins cooperate with the replication fork to faithfully restore H3K27me3 domains during DNA replication, which is discussed in-depth in a recent review on the subject (Borg et al. 2021a). Self-sustaining read–write mechanisms are thus conserved in animals and plants, which serve to mediate the stable epigenetic inheritance of transcriptional states and cellular identity from cell-to-cell.
Epigenetic silencing predates the origins of the plant kingdom
Our discussion will begin at the phylogenetic base of the Archaeplastida (or plant kingdom), the origins of which are traced to a primary photosynthetic endosymbiosis between a cyanobacterium and a eukaryotic host ~ 900 million years ago (Shih and Matzke 2013). The plant kingdom is represented by modern-day glaucophyte algae, red algae (or rhodophytes) and the Viridiplantae (green algae plus land plants) (Fig. 2). Algae is thus an encompassing term for a large group of diverse photosynthetic organisms from multiple eukaryotic clades, including stramenopiles like diatoms and brown algae. The concept of an alternation of generations can be misleading when applied to algae as, unlike land plants, there is often no fixed or regular alternation between two phases (John 1994). Interestingly, the term ‘alternation of generations’ arose from translation of the German term Generationwechsel—or change of generation—which is more appropriate to describe the non-obligate transitions observed in algae (John 1994). The term ‘life history’ has thus been proposed instead of ‘life cycle’ to more accurately reflect these complex changes in algal morphology and ploidy (John 1994).

Co-evolution of life history and epigenetic silencing in plants and algae. Summary of the most predominant life history features and prevalent role played by DNA, H3K9 and H3K27 methylation in the major lineages of plants and algae. A key is provided below to denote what each circle in the diagram represents. Features absent in a particular lineage lack a circle. The summary represents the most salient features garnered in this review and is by no means exhaustive. The phylogenetic tree was constructed using TimeTree (Kumar et al. 2017) with the following representative species: Phaeodactylum tricornutum (Diatoms), Ectocarpus siliculosus (Brown algae), Cyanidioschyzon merolae (Red algae), Chlamydomonas reinhardtii (Green algae), Marchantia polymorpha (Bryophytes), Ceratopteris richardii (Ferns), Picea abies (Gymnosperms) and Arabidopsis thaliana (Angiosperms)
Glaucophyte algae reproduce asexually through the formation of spores, with no sexual mode of reproduction reported thus far (Lopez et al. 2015). Red algae (or rhodophytes) form the largest group of mostly marine algae, some of which have high commercial value in agronomical and pharmaceutical industries. The life histories of red algae are some of the most complex known in living organisms, which is often haplo-diplontic but distinct in that triphasic life forms are expressed (Fig. 1) (Searles 1980). How this complex life history is controlled in red algae is unclear but a couple of studies have revealed that they harbor a functioning epigenetic machinery. For example, the red seaweed Gracilariopsis chorda is reported to have around 8% of its genome methylated (Lee et al. 2018). Much of this methylation is heavily biased to non-CG dinucleotides and is distributed equally between TEs, genes and promoters. Methylated TEs are also associated with small RNA, suggesting that RdDM might facilitate TE silencing in a manner similar to plants. The presence of H3K9 methylation and its potential synergy with DNA methylation in red algae is unknown (Fig. 2). Interestingly, at least half of all repetitive elements in the genome of the unicellular red alga Cyanidioschyzon merolae are marked with H3K27me3 (Mikulski et al. 2017). A small number (4%) of protein-coding genes are also marked with H3K27me3, which like H3K27me3-marked TEs, have low-to-undetectable levels of gene expression. Thus, in addition to small RNA-associated DNA methylation, H3K27me3 in red algae predominantly marks transcriptionally silent repetitive elements (Fig. 2).
Green algae are classified into two main groups—the Chlorophyte and Charophyte algae (Stewart and Mattox 1975). Chlamydomonas is a unicellular Chlorophyte and represents the most studied algal model organism. Aside from biotechnological applications, the phylogenetic position of Chlamydomonas makes it an ideal model organism to address the evolution of multicellularity in the plant kingdom. Compared to its green multicellular relatives, Chlamydomonas has a relatively simple haplontic life cycle with no distinct sporophytic generation (Fig. 1). The diploid zygote could thus be regarded as a sporophyte-like generation in Chlamydomonas and related unicellular algae, which undergoes meiosis to regenerate more haploid individuals, resulting in an alternation between haploid and diploid unicellular generations (Fig. 2). Despite this relatively simple life cycle, Chlamydomonas expresses a significant number of gamete-specific and zygote-specific genes, suggesting that silencing mechanisms exist to developmentally regulate these genes during its life cycle (Lopez et al. 2015).
DNA methyltransferase enzymes have ancient origins in green algae, including DNMT1 orthologs (Pei et al. 2019), while DNA methylation is detectable albeit at low (~ 1%) levels in Chlamydomonas and the related chlorophyte Volvox carteri (Babinger et al. 2007; Lopez et al. 2015). DNA methylation is confined to repeats in Chlamydomonas and remains stable across its life cycle (Lopez et al. 2015) but is altered during environmental adaptation (Kronholm et al. 2017), while it is linked with transgene silencing in Volvox (Babinger et al. 2001). Epigenetic silencing of exogenous transgenes is also reported in Chlamydomonas, which has been used to isolate causal silencing pathways (Jeong et al. 2002; Neupert et al. 2020). One such screen identified SET3p, a protein homologous to the Arabidopsis SUVH family that functions in vitro as a specific H3K9 mono-methyltransferase (Casas-Mollano et al. 2007). Knock-down expression of SET3p globally reduces H3K9me1 levels, including at the transgenic tandemly repeated arrays that are transcriptionally re-activated in its absence. The impact of SET3p knock-down on Chlamydomonas development was not reported, nor is it known whether H3K9me1 silences loci beyond repetitive elements. H3K9me3 is also detectable in Chlamydomonas but its deposition profile and catalysis still remain unknown (Casas-Mollano et al. 2007). Thus, H3K9 methylation was present in the ancestral lineage that gave rise to plants (Fig. 2), which was adapted during plant evolution to employ H3K9me2 as the dominant silencing modification of constitutive heterochromatin.
Phylogenetic identification suggest that PRC2 components were present in the common ancestor of red and green algae (Shaver et al. 2010; Huang et al. 2017b, 2019). While H3K27me3 is not detected in Chlamydomonas (Shaver et al. 2010; Mikulski et al. 2017; Khan et al. 2018), H3K27me1 is substantial and is catalyzed by a SET domain H3K27 methyltransferase homologous to E(z) (Shaver et al. 2010). These H3K27me1 marks co-occur on the same histone H3 tails that carry H3K4me1 (Khan et al. 2018), the latter of which appears to play an unusual transcriptional silencing role in Chlamydomonas (van Dijk et al. 2005). Knock-out of Chlamydomonas E(z) causes a global enrichment of active H3K4me3 marks and derepresses transgenes and transposons (Shaver et al. 2010), which interestingly phenocopies the loss of H3K4me1 (van Dijk et al. 2005). The tail of Chlamydomonas histone H3 contains an S28T mutation and deletion of residue A29 (Shaver et al. 2010), which might explain the altered mono-rather than tri-methylation at K27. This is reminiscent of H3K27me1 in Arabidopsis, which also silences TEs and is selectively deposited on histone H3.1 due to an A31T mutation (Jacob et al. 2009, 2014). Thus, a canonical role of PRC2-mediated silencing through deposition of H3K27me3 appears to be absent in Chlamydomonas, but is represented by a derived or perhaps ancestral system of transcriptional silencing that involves co-operation between H3K4me1 and H3K27me1.
DNA methylation is essential for bryophyte development
The transition from water to land was a major event in plant evolution that profoundly impacted terrestrial landscapes on earth (Dahl and Arens 2020). The origin of early land plants traces to ancestors of aquatic green algae, with the Zygnematophyceae ‘pond scum’ thought of as the most likely sister group of land plants (Zhong et al. 2015; Puttick et al. 2018). The closest descendants of this land colonizing lineage were a diverse group of non-vascular land plants called the bryophytes, which are represented by modern-day mosses, liverworts and hornworts. The bryophyte life cycle involves obligate transitions between a dominant haploid vegetative gametophyte and a relatively short-lived diploid sporophyte (Hisanaga et al. 2019). The obligate transition in their life cycle, together with their ancient phylogenetic origins, makes bryophytes a valuable model system to explore the control and evolution of life history transitions.
The two most intensely studied bryophyte model organisms are the moss Physcomitrella patens and the liverwort Marchantia polymorpha (Rensing et al. 2008; Bowman et al. 2017), both of which harbor substantial levels of CG, CHG and CHH methylation in their genome (Schmid et al. 2018; Domb et al. 2021). While the DNA methylation machinery is largely conserved with angiosperms, CHH methylation is instead catalyzed by two DNMT3-type methyltransferases homologous to mammalian de novo methyltransferases (Cao et al. 2000; Yaari et al. 2019). The DNMT3 family became extinct in angiosperms after likely giving rise to the DRM family through rearrangement of the DNMT3 methyltransferase domain (Cao et al. 2000). DNA methylation accumulates heavily on TEs and major satellite repeats in bryophytes but, unlike seed plants, appears to be excluded from ribosomal DNA (Matyášek et al. 2019). The silencing role of DNA methylation is conserved in Physcomitrella, with non-CG methylation in particular playing an important role in the transcriptional repression of not only TEs but also several protein-coding genes (Fig. 2) (Domb et al. 2021).
CG methylation is maintained by orthologs of MET1, which are essential for normal gametophyte development and sexual reproduction in Marchantia (Ikeda et al. 2018). In contrast, met1 gametophytes develop normally in Physcomitrella but fail to undergo sporophyte formation, although it is unclear whether this is caused by defects in gametogenesis, fertilization or sporophyte development (Yaari et al. 2015). However, severe gametophytic defects in Physcomitrella are observed in the presence of methyltransferase inhibitors (Malik et al. 2012) and in mutants of the single CMT3-type CHG methyltransferase, which incidentally also fails to form a sporophyte (Noy-Malka et al. 2014). Loss of CHH methylation in DNMT3 mutants, on the other hand, appears to have little impact on Physcomitrella development since these mutants are completely viable and develop normally (Yaari et al. 2019). Interestingly, DNA methylation undergoes extensive reprogramming across the life cycle in Marchantia (Schmid et al. 2018). This involves substantial gains in CHH methylation at TEs and repeats during sporophyte development, as well as enriched CG and CHG methylation on gene bodies and their flanking regions within sexual organs and sperm (Schmid et al. 2018). Marchantia thus undergoes at least two waves of DNA methylation reprogramming during its life cycle, which occurs at both TEs and protein-coding genes. A detailed examination of the loci targeted during these reprogramming cycles, together with further profiling of mutant methylomes, might offer clearer insight into how DNA methylation impacts the transition between haploid and diploid life.
What is particularly striking is the dramatic hyper-methylation observed in Marchantia sperm (Schmid et al. 2018), where at least half of all genes accumulate 4-methylcytosine (4mC) at CG dinucleotides (Walker et al. 2021), a form of methylation previously thought to be restricted only to prokaryotes (Ye et al. 2017; O’Brown et al. 2019). 4mC was shown to be catalyzed by two novel sperm-specific DNA methyltransferases, MpDN4MT1a/b, which are essential for sperm transcription and viability (Walker et al. 2021). How prevalent 4mC is across the Marchantia life cycle and other eukaryotes is unclear but its discovery raises exciting questions, not least how this DNA modification might influence genomic activity differently from 5mC. Like animals, Marchantia produce motile sperm and densely package sperm chromatin with protamines (Reynolds and Wolfe 1978; Borg and Berger 2015), suggesting that hyper-methylation of the paternal genome might contribute to chromatin condensation and/or influences events during early embryogenesis.
PRC2 represses the sporophyte transition in bryophytes
As we have discussed, PRC2 is present in unicellular red and green algae, such that it was likely inherited by bryophytes during evolution of the green lineage (Fig. 2) (Huang et al. 2017b; Schubert 2019). Deletion or knock-down of E(z) orthologs causes precocious formation of sporophyte-like bodies in Physcomitrella gametophytes (Okano et al. 2009; Pereman et al. 2016) and gametophyte lethality in Marchantia, respectively (Flores-Sandoval et al. 2016). Similarly, loss of the FIE ortholog, which probably tethers PRC2 to H3K27me3 as in animals (Margueron et al. 2009), negatively impacts gametophyte development and causes the formation of sporophyte-like structures in the absence of fertilization (Mosquna et al. 2009). Other ‘readers’ of H3K27me3 appear to have arisen multiple times during evolution (Schubert 2019), including the chromodomain protein LHP1 and two bromo-adjacent homology (BAH)–plant homeodomain (PHD) proteins in Arabidopsis (Turck et al. 2007; Zhang et al. 2007; Li et al. 2018; Yang et al. 2018). These H3K27me3 readers appear to have ancient origins in land plants (Berke and Snel 2015; Huang et al. 2019) but also in filamentous fungi like Neurospora crassa (Wiles et al. 2020). A loss-of-function lhp1 mutant in Physcomitrella reportedly displays pleiotropic defects in the gametophyte, although latter stages of development were not reported in this study (Dangwal et al. 2014).
Among H3K27me3-silenced genes in bryophytes are the evolutionary conserved BELL and KNOX TALE-homeodomain transcription factors (TF), which interact to activate diploid gene expression and zygote formation in Chlamydomonas and Marchantia (Lee et al. 2008; Widiez et al. 2014; Horst and Reski 2016; Horst et al. 2016; Dierschke et al. 2020). Interestingly, ectopic overexpression of BELL1 in Physcomitrella induces embryo and sporophyte formation in the absence of fertilization (Horst et al. 2016). This likely explains the sporophyte-like features of PRC2 mutant gametophytes since BELL1 has been shown to de-repressed in these mutants (Pereman et al. 2016). The ancient KNOX-BELL program was thus likely co-opted by PRC2 to control the gametophyte-to-sporophyte transition during land plant evolution (Fig. 2). In addition to PRC2-mediated repression, the Physcomitrella sporophytic program is reinforced by KNOX2 class TFs that repress the haploid program in the diploid plant body (Sakakibara et al. 2013). How KNOX, BELL and other H3K27me3-silenced genes are reprogrammed during the earliest phases of the sporophyte transition is not known. H3K27me3 is lost at hundreds of protein-coding genes during the transition from juvenile to mature gametophytes in Physcomitrella (Widiez et al. 2014), further implying the presence of mechanisms that reprogram H3K27me3 during development. In both animals and angiosperms, H3K27me3 is extensively reprogrammed during reproductive development through histone exchange and/or active demethylation mechanisms (Hajkova 2011; Zheng et al. 2016; Borg et al. 2020). It is thus of interest to explore mechanisms of H3K27me3 reprogramming in bryophytes since these might also contribute to the control of alternating generations.
The origin of PRC2 lies in the silencing of transposons
A striking difference in the epigenetic landscape of Physcomitrella and Marchantia is the heterochromatic nature of TEs. Like angiosperms, TEs in Physcomitrella are largely marked by constitutive H3K9me2-marked heterochromatin, whereas almost 40% are marked exclusively with H3K27me3 in Marchantia (Fig. 2) (Widiez et al. 2014; Montgomery et al. 2020). It is worth noting that these chromatin profiles were generated from gametophytic tissue, so whether the composition of heterochromatin is altered in the sporophyte is unclear. Changes to heterochromatin are known to occur in the angiosperm female gametophyte, where the reduction in DNA and H3K9 methylation coincides with a redistribution of H3K27me3 to a large proportion of TEs (Weinhofer et al. 2010). Accumulation of H3K27me3 at TEs also occurs in mutants affecting constitutive heterochromatin in fungi, animals and even plants (Reddington et al. 2013; Jamieson et al. 2016; Rougée et al. 2019). We have already highlighted how transposons and repeats are marked by H3K27me3 in unicellular red algae and green algae (Fig. 2), which also extends to diatoms and ciliates (Veluchamy et al. 2015; Mikulski et al. 2017; Frapporti et al. 2019). H3K27me3 is also co-deposited with H3K9me3 at TEs in Paramecium tetraurelia by a PRC2-like SET domain methyltransferase (Frapporti et al. 2019). These observations suggest that H3K27me3 has an ancestral role in the silencing of transposable elements, perhaps even in common with H3K9 methylation (Henikoff and Ahmad 2020). Marchantia is particularly compelling since TE silencing is partitioned between either H3K27me3 or DNA-H3K9 methylation (Montgomery et al. 2020). The redundant function of both pathways went on to diverge in a division of labor during plant evolution, with H3K27me3 becoming dedicated to repressing gene expression and DNA-H3K9 methylation dominating the silencing of TEs.
Ferns began the transition to a dominant sporophyte generation
After bryophytes colonized terrestrial habitats, they were challenged by a new group of plants called the pteridophytes, which developed a complex vascular system. Ferns and fern-like plants are modern-day descendants of the earliest vascular plants, which went on to dominate the flora on earth during the Carboniferous period (Dimichele and Phillips 2002). The life history of ferns is reversed from that of bryophytes in that the gametophyte generation is highly reduced compared to the vegetative sporophyte (Banks 1999). Haploid spores are produced by sporangia on the underside of the leafy fronds of the fern, which then germinate into tiny free-living gametophytes. These gametophytes will differentiate the gametes, which eventually fuse to form the embryo and re-initiate sporophyte development. In Ceratopteris richardii and many other ferns, the sex of gametophytes is determined by a male-inducing pheromone (Warne and Hickok 1991), which interestingly stimulates the transcription of several epigenetic modifiers, including H3K9, H3K27 and DNA methyltransferases (Atallah et al. 2018). Heritable epigenetic silencing through DNA methylation also appears to be functional in related ferns like Adiantum capillus-veneris (Tsuboi et al. 2012). Thus, ferns appear to have inherited an epigenetic machinery from a common ancestor with their sister bryophyte lineage, but how this might impact life history transitions remains to be seen (Fig. 2).
Adaptive traits in gymnosperms indicate an epigenetic-based memory
The domination of ferns and fern-like plants was eventually overtaken by seed plants, which innovated hardy seeds to protect the embryonic sporophyte and aid its dispersal over long distances. Similarly, the innovation of microscopic gametophytes also assisted gene flow over long distances through adaptive mechanisms of pollen dispersal (Levin and Kerster 1974). While the female gametophyte remains buried within the tissues of the sporophyte and forms the embryo sac, the male gametophyte develops into a free-living pollen grain that delivers sperm for fertilization (Rudall 2006b; Rudall and Bateman 2007b). Thus, unlike bryophytes and ferns, seed plants no longer relied on water and flagellate sperm for reproduction, breaking one of the last remaining links with their aquatic cousins.
Gymnosperms represent a major group of seed-producing plants, most of which are large conifer trees with cones that bear spore-producing sporangia. Norway spruce (Picea abies) is one such common conifer species that has important ecological and economic value in European forests. Norway spruce harbors diverse histone methylation marks (Fuchs et al. 2008), undergoes dynamic changes in H3K27me3 and DNA methylation during embryonic tissue culture (Nakamura et al. 2020), exhibits soma clonal methylome variation (Ausin et al. 2016; Heer et al. 2018) and, like angiosperms, expresses a distinct population of small RNAs in pollen (Nakamura et al. 2019). Interestingly, breeders have noted adaptive traits in Norway spruce where epigenetic events early in development (i.e., post-meiotic megagametogenesis and seed maturation) can determine how a tree behaves and grows years later, thus allowing the expression of an altered phenology when grown in a non-native environment (Yakovlev et al. 2012). Strikingly, these adaptive traits have a strong parental effect, since differences in day length and temperature during female flowering has an effect on progeny performance while that during pollen formation does not (Johnsen et al. 1996, 2005). Maternal transmission of adaptive potential is consistent with that reported in angiosperms (Wibowo et al. 2016; Luo et al. 2020), which incidentally also extensively reprogram paternal epigenetic memory (Borg et al. 2020). This implies some form of differential epigenetic reprogramming in male and female gymnosperm gametophytes, which might explain this parentally biased adaptation to different climates.
A pre-meitoic wave of reprogramming precedes the diploid-to-haploid transition in angiosperms
The angiosperms (or flowering plants) dominate the terrestrial vegetation on earth and thus represent the most successful and diverse group of land plants. As seed plants, angiosperms share many of the innovative attributes that arose in gymnosperms, with multiple other drivers contributing to their prominence (Augusto et al. 2014). The haploid transition begins once diploid spore mother cells undergo meiosis to produce the haploid male microspore and the female megaspore (Baroux and Autran 2015). Megaspore mother cells (MMCs) are specified from nucellar cells of the ovule primordium, while pollen mother cells (PMCs) develop from sporogenous cells of the anther. Strikingly, both the MMC and PMC in Arabidopsis are characterized by several cytological changes at the level of chromatin, including a general decondensed chromatin state, the depletion of linker histone H1, the eviction of replicative histone H3.1 and a drastic reduction in the levels of H3K27me3 (She et al. 2013; She and Baroux 2015; Hernandez-Lagana and Autran 2020). In the PMC, these changes in chromatin are accompanied by a modest RdDM-dependent increase in CHH methylation, which regulates the RNA splicing of genes required for normal progression through meiosis (Walker et al. 2018). Thus, diploid spore mother cells undergo reprogramming both at the level of histone and DNA methylation, which likely begins to establish an epigenetic landscape that distinguishes them from surrounding diploid sporophytic cells.
Epigenetic reprogramming controls seed development and speciation in angiosperms
The angiosperm life cycle is largely shared with gymnosperms, with the important difference being that the microscopic gametophytes give rise to two gametes that participate in a double fertilization event. The female gametophytes develop into an embryo sac containing the egg and central cell, while the male gametophytes develop into pollen grains containing two sperm within a companion vegetative cell. Pollen delivery of the sperm to the embryo sac initiates seed development, where fertilization of the egg and central cell forms the embryo and endosperm, respectively. Many of the first epigenetic mutants in Arabidopsis were isolated based on phenotypes that negatively affected seed development. Maternal inheritance of fertilization-independent seed (fis) mutations causes the central cell to precociously undergo endosperm development in the absence of fertilization, which abort prematurely even if the egg is fertilized (Ohad et al. 1996; Chaudhury et al. 1997; Köhler and Makarevich 2006). These fis mutations were mapped to MEDEA (MEA) (Grossniklaus et al. 1998; Kiyosue et al. 1999), FERTILIZATION‐INDEPENDENT ENDOSPERM (FIE) (Ohad et al. 1999), FIS2 (Luo et al. 1999) and MULTI‐COPY SUPPRESSOR OF IRA1 (MSI1) (Köhler et al. 2003), all of which encode core subunits of the PRC2 complex. H3K27me3-mediated repression thus plays a key role during female gametophyte development and likely stabilizes cellular fate in the central cell and developing endosperm.
MEA and FIS2 are only expressed from maternal alleles in the endosperm, while paternal alleles remain silenced throughout seed development (Köhler and Makarevich 2006). This phenomenon, called genomic imprinting, is a characteristic feature of gene expression in the endosperm (Gehring and Satyaki 2017). Several maternally and paternally expressed imprinted genes (MEGs and PEGs) have been identified in Arabidopsis and rice (Gehring et al. 2009; Moreno-Romero et al. 2019; Borg et al. 2020), whose monoallelic expression arises from the differential retention of H3K27me3 or DNA methylation during male and female gametogenesis (Gehring et al. 2009; Moreno-Romero et al. 2019; Borg et al. 2020). The central cell is homodiploid in most angiosperms due to the fusion of two nuclei during female gametophyte development (Slotkin et al. 2009; Schoft et al. 2009; Mérai et al. 2014; He et al. 2019). Karyogamy with haploid sperm thus results in an endosperm with double the maternal genome dosage, which is essential since increasing paternal dosage leads to seed abortion (Zhang 2016). This dosage-sensitivity is caused by the increased expression of PEGs, with at least four PEGs demonstrated to cause seed lethality in Arabidopsis (Kradolfer et al. 2013; Wolff et al. 2015; Huang et al. 2017a; Wang et al. 2018). This forms the basis for an endosperm-based post-zygotic hybridization barrier called the ‘triploid block,’ which is proposed to drive speciation in angiosperms (Köhler et al. 2010; Lafon-Placette et al. 2017; Tonosaki et al. 2018).
The central cell also undergoes epigenetic reprogramming of constitutive heterochromatin through the depletion of H3K9me2 and active DNA demethylation (Hsieh et al. 2009; Pillot et al. 2010; Ibarra et al. 2012; Park et al. 2016). The latter is controlled by DEMETER (DME), a DNA glycosylase that demethylates cytosines at several genes flanked by transposons and repeats, including MEGs like MEA (Choi et al. 2002; Gehring et al. 2006; Park et al. 2017). DNA demethylation appears to be critical for endosperm development since, like fis mutants, the maternal inheritance of dme mutations severely impacts seed viability (Choi et al. 2002; Gehring et al. 2006). Interestingly, two PEGs that induce the triploid block encode proteins involved in H3K9me2 homeostasis, namely ADMETOS (ADM) and SUVH7 (Kradolfer et al. 2013; Wolff et al. 2015). ADM induces the triploid block through the ectopic accumulation of H3K9me2 at AT-rich TEs, which deregulates the expression of neighboring genes in the developing endosperm (Kradolfer et al. 2013). These observations suggest that the loss of H3K9me2 is involved in specifying aspects of central cell fate, which in turn facilitates the development of the triploid endosperm after fertilization.
The loss of DNA-H3K9 methylation rewires gene regulatory networks during Arabidopsis pollen development
Like the central cell, the pollen vegetative cell also undergoes epigenetic reprogramming of DNA-H3K9 methylation (Schoft et al. 2009; Ibarra et al. 2012). This appears to be more extensive than in the central cell since pericentromeric heterochromatin is dramatically decondensed, resulting in highly diffuse chromatin in the vegetative cell nucleus (VN) (Borg and Berger 2015). This epigenetic reconfiguration is caused by several mechanisms, namely the depletion of the heterochromatin remodeller DECREASE IN DNA METHYLATION 1 (DDM1) and linker histone H1, the loss of H3K9me2 and the unloading of centromeric H3 (Slotkin et al. 2009; Schoft et al. 2009; Mérai et al. 2014; He et al. 2019). Heterochromatin decondensation facilitates active DNA demethylation by DME, a process that appears to be important for pollen germination and male fertility, at least in certain Arabidopsis ecotypes (Schoft et al. 2011; He et al. 2019). As a result, the loss of constitutive heterochromatin identity in the VN re-activates a handful of TEs that stimulate the production of epigenetically activated small RNAs (easiRNAs) (Slotkin et al. 2009; Calarco et al. 2012; Borges et al. 2018; Wang et al. 2020).
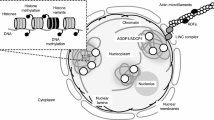
Strikingly, a large proportion of the genomic regions targeted for demethylation by DME specifically gain chromatin accessibility in the VN (Borg et al. 2021b). This occurs within regions that not only stimulate easiRNA production but that also lie in the vicinity of VN-specific protein-coding genes, which importantly are silenced with DNA-H3K9 methylation in the sporophyte (Borg et al. 2021b). These regions are enriched for the predicted binding site of several VN-expressed TFs, many of which are unable to bind to their cognate binding sites when DNA is methylated (O’Malley et al. 2016), such that DME-mediated demethylation likely licenses their binding (Fig. 3). Consistently, DME is directly required for the expression of at least 27 of these genes, several of which have importantly been shown to play a direct role in controlling pollen tube growth (Borg et al. 2021b; Khouider et al. 2021). Thus, the reprogramming of DNA-H3K9 methylation helps rewire haploid gene expression by exposing TF binding sites normally repressed during sporophytic life (Fig. 3).

Loss of DNA-H3K9 methylation patterns the Arabidopsis male gametophyte. Model summarizing how the epigenetic reprogramming of DNA-H3K9 methylation in the VN activates the male gametophytic program. In the sporophyte generation, a subset of pollen-expressed genes remains silenced with constitutive heterochromatin. During pollen development, the VN undergoes extensive reprogramming to disassemble constitutive heterochromatin, which includes the loss of H3K9me2 (yellow dots) and depletion of linker histone H1 (gray ovals). The DNA glycosylase DEMETER (gray pacman) is now able to actively demethylate the high levels of DNA methylation (red dots) known to accumulate in these heterochromatic regions. This exposes cis-regulatory elements (green lines) in the vicinity of pollen-expressed genes normally silenced during sporophytic life, allowing methylation-sensitive TFs to bind and activate their expression. The loss of DNA-H3K9 methylation thus leads to transcriptional reprogramming and facilitates the sporophyte-to-gametophyte transition
The disassembly of pericentromeric heterochromatin in the VN also impacts the regulation of ribosomal RNA in pollen. Ribosomal DNA (rDNA) genes are clustered at two nucleolar organizing regions (NORs) in Arabidopsis (Copenhaver and Pikaard 1996; Rabanal et al. 2017). Both NORs associate with centromeric heterochromatin and lie externally along the nucleolus in leaf nuclei, where only one NOR undergoes active transcription to produce ribosomal RNA (Mérai et al. 2014; Rabanal et al. 2017). In pollen, these NORs coalesce and become internalized in the nucleolus of the VN, which also correlates with the re-activation of rDNA variants from the silenced NOR (Mérai et al. 2014). This suggests that, in addition to pollen tube genes, rDNA loci that are normally silenced during sporophytic life become re-activated in pollen. This is likely key for increasing translational potential of the rapidly growing pollen tube. Taken together, these mechanistic insights refine our understanding of epigenetic reprogramming in the VN and suggests that its primary role is to rewire transcription and facilitate the male sporophyte-to-gametophyte transition in angiosperms (Fig. 4).

Epigenetic reprogramming in the male gametophyte guides the alternation of generations in Arabidopsis. The haploid epigenome undergoes extensive and differential epigenetic reprogramming during male gametophyte development. In the vegetative cell, the loss of DNA methylation and H3K9me2 relieves transcriptional silencing over pollen-expressed genes normally silenced in the diploid sporophyte to activate genes required for pollen tube growth and sperm delivery. In sperm, the loss of H3K27me3 relieves transcriptional silencing over both sperm-specific genes and master regulators of early embryogenesis. Thus, the loss of DNA-H3K9 methylation facilitates the sporophyte-to-gametophyte transition in the vegetative cell, while the loss of H3K27me3 primes the paternal genome for the gametophyte-to-sporophyte transition after fertilization
Paternal resetting of H3K27me3 primes sporophyte development in Arabidopsis
In contrast to the vegetative cell, the sperm cells undergo a distinct wave of epigenetic reprogramming that drives the genome-wide loss of H3K27me3 in angiosperms (Sano and Tanaka 2010; Houben et al. 2011; Borg et al. 2020). This resetting event involves the concerted action of three mechanisms in Arabidopsis, namely (1) the loss of PRC2 activity and capacity to write H3K27me3 (2) active demethylation by Jumonji-C family H3K27 demethylases and (3) the sperm-specific deposition of histone H3.10, which is immune to K27 methylation (Borg et al. 2020). Almost 50% of the genes specifically expressed in sperm are silenced with H3K27me3 in the sporophyte, such that the loss of H3K27me3 facilitates their transcription during spermatogenesis. Strikingly, several H3K27me3-marked genes in the sporophyte also include master regulators of embryogenesis like BABY BOOM (BBM) and LEAFY COTYLEDON 1 (LEC1) (Horstman et al. 2017; Khanday et al. 2019). In sperm, the loss of H3K27me3 as these loci coincides with increased chromatin accessibility and accumulation of active H3K4me3 modifications at promoter regions, which occurs in a pattern that predicts gene expression during earliest phases of embryogenesis (Borg et al. 2020, 2021b).
Paternal resetting of H3K27me3 thus primes the paternal genome for the sporophyte transition and has the potential to impact transcription in the early zygote. Nevertheless, these effects might be highly transient since paternal chromatin is rapidly reprogrammed in the zygote after fertilization (Ingouff et al. 2010). In contrast, paternally derived histones are passively diluted over several rounds of DNA replication in the endosperm (Ingouff et al. 2007), which might provide a longer window for histone-based imprints to act. Interestingly, unlike MEGs, many PEG loci are also transcriptionally primed with open chromatin and H3K4me3 in sperm (Borg et al. 2020, 2021b), providing correlative evidence for how paternal chromatin potentially impacts gene expression after fertilization. Mechanistically, pre-configured accessible chromatin could help elicit the binding of maternally derived TFs or perhaps already signifies a paternal contribution of pre-bound TFs at fertilization. In summary, the reprogramming of H3K27me3-silenced states in the male gametophyte generation orchestrates the rewiring of transcription to facilitate the haploid-to-diploid transition in angiosperms (Fig. 4).
Diatoms share a chimeric epigenetic landscape with animals and plants
Stramenopiles (or heterokonts) are a monophyletic group that arose independently from Archaeplastida through a secondary endosymbiosis event involving the capture of a red alga. They include diverse unicellular algae such as diatoms and water molds (or oomycetes) but also multicellular species of brown algae. Diatoms are essential plankton components of aquatic ecosystems that are famed for their dazzling and elaborate silica cell walls. Like animals, the diatom life cycle is diplontic with the haploid phase reduced down to the gametes (Fig. 1). Methylome profiling in several different diatom species has revealed generally low levels of DNA methylation with some exceptions ranging as high as ~ 60% (Jarvis et al. 1992; Veluchamy et al. 2013; Huff and Zilberman 2014; Traller et al. 2016). Much of this methylation is restricted to TEs and a few genes, whereas intergenic regions are generally devoid of methylation. The localized enrichment of high levels of DNA methylation in the model diatom Phaeodactylum tricornutum suggest deep links with chromatin structure, which is dynamically altered with transcription in response to environmental changes (Veluchamy et al. 2013). DNMT1 symmetric methyltransferases are absent in diatoms (Zemach and Zilberman 2010), with CG methylation likely deposited by DNMT5 methyltransferases known to be active in other CG-methylated species lacking DNMT1 (Huff and Zilberman 2014).
Phaeodactylum also harbors a wide repertoire of histone modifications (Veluchamy et al. 2015). H3K27me3 covers about 14% of the Phaeodactylum genome, which is unusually high but coherent with its high load of H3K27me3-marked TE insertions. The fraction of H3K27me3-marked genes in Phaeodactylum is altered as morphotypes undergo changes in cell shape, with an E(z) knock-out abolishing these elaborate shapes altogether (Zhao et al. 2021). These findings highlight a role of PRC2 in developmental regulation and cell identity in unicellular species that may be more ancient than previously appreciated (Zhao et al. 2021). Several forms of H3K9 methylation are also found in Phaeodactylum, with the relevant marked loci globally anti-correlated with transcription (Veluchamy et al. 2015). Like Paramecium (Frapporti et al. 2019a), H3K9 methylation in Phaeodactylum might be under control of an E(z) homolog since no other H3K9 methyltransferases are present while H3K9 and H3K27 methylation largely co-occur. Thus, diatoms have a chimeric epigenetic landscape that shares both animal and plant features where H3K9me2, H3K27me3 and DNA methylation all participate in the repression of TE activity.
Life history transitions in brown algae involves independent regulatory controls
Brown algae (or phaeophytes) are a large group of multicellular eukaryotes with diverse morphology ranging from tiny species like Ectocarpus siliculosus to 50-m-long giant kelps like Macrocystis pyrifera. As plants do on land, brown algae play fundamental ecological roles in supporting coastal marine ecosystems and have direct economic impact in food harvest, commercial extracts and marine fouling (Bringloe et al. 2020). Brown algae are haplo-diplontic organisms with the exception of the Fucales order which, like animals, have a diplontic life cycle (Fig. 1) (Cock et al. 2014). Developmentally, brown algae are fascinating in that they display a dazzling variety of life histories across the entire clade (Heesch et al. 2019). The transition between haploid and diploid phases is also often non-obligate in brown algae. For example, gametes in the model brown algae Ectocarpus can initiate the sporophyte program without fertilization, developing with or without endoreplication to become either diploid or haploid parthenosporophytes (Müller 1966; Bothwell et al. 2010). Ectocarpus can thus transition through a complete life cycle with the same genes and ploidy without fertilization, making it a compelling case study of the alternation of generations in brown algae.
Although they share a common ancestor with diatoms, neither DNA methylation nor its deposition machinery is detected in Ectocarpus (Cock et al. 2010). Ectocarpus has a relative simple morphology with only a few different cell types, which contrasts with the complex tissues and different cell types of giant kelps (Bringloe et al. 2020). DNA methylation has been reported in the kelp Saccharina japonica, albeit at very low (1.4%) levels (Fan et al. 2020). Much of this methylation appears to occur in a CHH context, anti-correlates with transcription, and targets a wide range of genomic features. The S. japonica genome only encodes a homolog of the tRNA methyltransferase DNMT2, leading to the supposition that these homologs methylate both RNA and DNA, although no biochemical evidence was presented to test this hypothesis. The unequivocal presence of methylated cytosines by mass spectrometry in S. japonica DNA is also lacking. Coupled with the trace amounts of DNA methylation reported and the lack of an obvious DNA methyltransferase, it is probably premature to conclude that brown algae harbor and regulate genome activity using DNA methylation.
Given how histone methylation regulates life cycle transitions in land plants, its role in brown algae is of key interest. While several histone marks are present in Ectocarpus, H3K27me3 is absent, while H3K9me2/3 is only detected at very low levels (Bourdareau et al. 2021). Consistently, no E(z) and PRC2 homologs have been identified in brown algae thus far, including Ectocarpus and S. japonica (Bourdareau et al. 2021). A homolog of EHMT2 (or G9a), which functions as a distinct H3K9 methyltransferase in animals, is present in Ectocarpus along with several known H3K9 demethylases (Maumus et al. 2011). Thus, while H3K27me3 is clearly absent, the status of H3K9 methylation remains inconclusive. Ectocarpus gametophytes and sporophytes are highly isomorphic, with the minimal changes in transcription reflected in the histone landscape across its life cycle (Bourdareau et al. 2021). It is worth noting that parthenosporophytes were assayed in this study, so sporophytes arising from fertilization might display a distinct profile, particularly since zygotic chromatin is known to undergo dramatic changes in plants and animals (Loppin et al. 2005; Bonnefoy et al. 2007; Ingouff et al. 2010). Thus, it remains to be resolved whether any form of epigenetic reprogramming is associated with the alternation of generations in Ectocarpus and whether this is common across the brown algal lineage.
Perspectives and future endeavors
Since its first discovery by Hofmeister over 170 years ago, a mesmerizing array of life history strategies have been described that mirror the increasing complexity of plants and algae. Epigenetic silencing provides a means to reshape differentiation and is a proposed driving force in the transition to multicellular life from the ancestral unicellular state (Gombar et al. 2014). Epigenetic silencing was likely present in the common ancestor of plants, where it primarily served as a genome defense system against TEs and exogenous retroviruses but then diverged into the control of gene expression (Fig. 2). With the advent of meiosis and fertilization, the stage would have been set for epigenetic regulation to begin diverging between the haploid and diploid phases of the life cycle, ultimately giving rise to the diverse life histories observed in modern-day plants and algae.
DNA methylation is a clear case in point. In green algae, DNA methylation is restricted to gene-poor regions and is largely static across the life cycle (Lopez et al. 2015). In bryophytes, DNA methylation also marks genes, is essential for development and undergoes dynamic changes during the life cycle (Noy-Malka et al. 2014; Yaari et al. 2015; Schmid et al. 2018; Ikeda et al. 2018). In Arabidopsis, DNA methylation silences a subset of gametophyte-specific genes together with H3K9 methylation in the sporophyte generation, which are both reprogrammed during male gametophyte development to license activation of the haploid program (Fig. 3) (Borg et al. 2021b; Khouider et al. 2021). DNA-H3K9 methylation is reconfigured similarly in the central cell of the female gametophyte (Pillot et al. 2010; Ibarra et al. 2012; Park et al. 2016), suggesting that this mechanism probably plays a general role in the sporophyte-to-gametophyte transition in angiosperms. DNA-H3K9 methylation is normally associated with TE silencing in flowering plants (Feng and Michaels 2015), but its role in the gametophyte highlights how it also represses developmental programs that govern lineage specification. TE insertions have rewired transcriptional networks during evolution in other eukaryotic lineages (Rebollo et al. 2012), and it is interesting that many male gametophyte-specific genes activated upon the loss of DNA-H3K9 methylation are associated with TEs (Borg et al. 2021b). TE activity could have thus altered the gametophytic program in ancestral land plants, providing fertile ground for regulatory mechanisms to evolve and control haploid–diploid transitions.
Unlike H3K9 methylation, PRC2-mediated silencing through H3K27me3 is largely dedicated to regulating growth and development in complex eukaryotes (Margueron and Reinberg 2011). However, as we have discussed, the ancestral role of PRC2 was also a genome defense system that evolved toward developmental regulation and cell identity (Fig. 2). PRC2 still functions today to repress TE activity in unicellular red and green algae (Mikulski et al. 2017) but also bryophytes (Montgomery et al. 2020). At some point in the evolution of early land plants, PRC2 began to silence gene expression, which included the BELL/KNOX system that regulates the diploid program in green algae and bryophytes (Lee et al. 2008; Widiez et al. 2014; Horst and Reski 2016; Horst et al. 2016; Dierschke et al. 2020). This scenario would have led to repression of the sporophyte program and extension of the gametophytic phase, which is still evident in modern-day bryophytes. In Arabidopsis, the loss of H3K27me3 in sperm might have a similar role, since this relieves silencing over several master regulators that initiate the sporophyte program in angiosperms (Horstman et al. 2017; Borg et al. 2020). Thus, studies of chromatin reprogramming during male gametophyte development suggest a model where programmed loss of H3K27me3 and DNA-H3K9 methylation act independently to facilitate life cycle transitions in Arabidopsis (Fig. 4). Further investigation of chromatin reprogramming in the female gametophyte and other flowering plant species will help determine how general the mechanisms operating in Arabidopsis pollen are for the gametophyte transition in angiosperms.
The independent evolution of the alternation of generations in the Stramenopile lineage raises questions about what is common or different about its molecular control. Brown algae display a complex array of life history strategies (Heesch et al. 2019), but the clear absence of PRC2 silencing already suggests that aspects of the sporophyte transition in plants cannot be generalized (Fig. 2). Moreover, the fact that PRC2 pathways are functional and essential for cell identity in diatoms but were lost in brown algae is further compelling and suggests that novel regulatory mechanisms could have emerged during Stramenopile evolution. Despite the differences in epigenetic control, two TALE-homeodomain TFs are known to promote sporophyte formation in Ectocarpus (Coelho et al. 2011a; Arun et al. 2019). This mode of regulation bears a striking and remarkable similarity with the BELL/KNOX system in the green lineage. The evolution and regulation of the life history transitions is thus clearly complex and likely involves both common and lineage-specific mechanisms. Future endeavors must expand across and beyond the green lineage to provide both a molecular and evolutionary understanding of the alternation of generations, which is what ultimately makes the developmental biology of plants and algae so fascinating.
References
Arimondo PB, Barberousse A, Pontarotti G (2019) The many faces of epigenetics. Epigenetics. https://doi.org/10.1080/15592294.2019.1595298
Arun A, Coelho SM, Peters AF et al (2019) Convergent recruitment of TALE homeodomain life cycle regulators to direct sporophyte development in land plants and brown algae. Elife. https://doi.org/10.7554/eLife.43101
Atallah NM, Vitek O, Gaiti F et al (2018) Sex determination in Ceratopteris richardii is accompanied by transcriptome changes that drive epigenetic reprogramming of the young gametophyte. G3: GEnes Genom Genet 8:10. https://doi.org/10.1534/g3.118.200292
Audergon PNCB, Catania S, Kagansky A et al (2015) Restricted epigenetic inheritance of H3K9 methylation. Science. https://doi.org/10.1126/science.1260638
Augusto L, Davies TJ, Delzon S, de Schrijver A (2014) The enigma of the rise of angiosperms: Can we untie the knot? Ecol Lett 17:1326–1338
Ausin I, Feng S, Yu C et al (2016) DNA methylome of the 20-gigabase Norway spruce genome. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1618019113
Babinger P, Kobl I, Mages W, Schmitt R (2001) A link between DNA methylation and epigenetic silencing in transgenic Volvox carteri. Nucleic Acids Res. https://doi.org/10.1093/nar/29.6.1261
Babinger P, Völkl R, Cakstina I et al (2007) Maintenance DNA methyltransferase (Met1) and silencing of CpG-methylated foreign DNA in Volvox carteri. Plant Mol Biol. https://doi.org/10.1007/s11103-006-9091-1
Banks JA (1999) Gametophyte development in ferns. Annu Rev Plant Biol. https://doi.org/10.1146/annurev.arplant.50.1.163
Baroux C, Autran D (2015) Chromatin dynamics during cellular differentiation in the female reproductive lineage of flowering plants. Plant J. https://doi.org/10.1111/tpj.12890
Bell PR (1989) The alternation of generations. Adv Bot Res 16:55–93. https://doi.org/10.1016/S0065-2296(08)60239-6
Berke L, Snel B (2015) The plant Polycomb repressive complex 1 (PRC1) existed in the ancestor of seed plants and has a complex duplication history. BMC Evol Biol. https://doi.org/10.1186/s12862-015-0319-z
Bewick AJ, Schmitz RJ (2017) Gene body DNA methylation in plants. Curr Opin Plant Biol 36:103–110
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21. https://doi.org/10.1101/gad.947102
Bird A (2007) Perceptions of epigenetics. Nature 447:396–398
Bonasio R, Tu S, Reinberg D (2010) Molecular signals of epigenetic states. Science 330:612–616
Bonnefoy E, Orsi GA, Couble P, Loppin B (2007) The essential role of Drosophila HIRA for de novo assembly of paternal chromatin at fertilization. PLoS Genet 3:1991–2006. https://doi.org/10.1371/journal.pgen.0030182
Borg M, Berger F (2015) Chromatin remodelling during male gametophyte development. Plant J 83:177–188. https://doi.org/10.1111/tpj.12856
Borg M, Jacob Y, Susaki D et al (2020) Targeted reprogramming of H3K27me3 resets epigenetic memory in plant paternal chromatin. Nat Cell Biol. https://doi.org/10.1038/s41556-020-0515-y
Borg M, Jiang D, Berger F (2021a) Histone variants take center stage in shaping the epigenome. Curr Opin Plant Biol 61:101991
Borg M, Papareddy RK, Dombey R et al (2021b) Epigenetic reprogramming rewires transcription during the alternation of generations in arabidopsis. Elife 10:10. https://doi.org/10.7554/eLife.61894
Borges F, Parent JS, van Ex F et al (2018) Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis. Nat Genet. https://doi.org/10.1038/s41588-017-0032-5
Bostick M, Jong KK, Estève PO et al (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. https://doi.org/10.1126/science.1147939
Bothwell JH, Marie D, Peters AF et al (2010) Role of endoreduplication and apomeiosis during parthenogenetic reproduction in the model brown alga Ectocarpus. New Phytol. https://doi.org/10.1111/j.1469-8137.2010.03357.x
Bourdareau S, Tirichine L, Lombard B et al (2021) Histone modifications during the life cycle of the brown alga Ectocarpus. Genome Biol. https://doi.org/10.1186/s13059-020-02216-8
Bowman JL, Kohchi T, Yamato KT et al (2017) Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell. https://doi.org/10.1016/j.cell.2017.09.030
Bringloe TT, Starko S, Wade RM et al (2020) Phylogeny and evolution of the brown algae. Crit Rev Plant Sci. https://doi.org/10.1080/07352689.2020.1787679
Calarco JP, Borges F, Donoghue MTA et al (2012) Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151:194–205. https://doi.org/10.1016/j.cell.2012.09.001
Cao X, Springer NM, Muszynski MG et al (2000) Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.97.9.4979
Casas-Mollano JA, van Dijk K, Eisenhart J, Cerutti H (2007) SET3p monomethylates histone H3 on lysine 9 and is required for the silencing of tandemly repeated transgenes in Chlamydomonas. Nucleic Acids Res. https://doi.org/10.1093/nar/gkl1149
Catania S, Dumesic PA, Pimentel H et al (2020) Evolutionary persistence of DNA methylation for millions of years after ancient loss of a de novo methyltransferase. Cell. https://doi.org/10.1016/j.cell.2019.12.012
Chaudhury AM, Ming L, Miller C et al (1997) Fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci USA 94:4223–4228. https://doi.org/10.1073/pnas.94.8.4223
Choi Y, Gehring M, Johnson L et al (2002) DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 110:33–42. https://doi.org/10.1016/S0092-8674(02)00807-3
Cock JM, Sterck L, Rouzé P et al (2010) The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature. https://doi.org/10.1038/nature09016
Cock JM, Godfroy O, Macaisne N et al (2014) Evolution and regulation of complex life cycles: a brown algal perspective. Curr Opin Plant Biol 17:1–6
Coelho SM, Godfroy O, Arun A et al (2011a) OUROBOROS is a master regulator of the gametophyte to sporophyte life cycle transition in the brown alga Ectocarpus. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1102274108
Coelho SM, Godfroy O, Arun A et al (2011b) Genetic regulation of life cycle transitions in the brown alga ectocarpus. Plant Signal Behav. https://doi.org/10.4161/psb.6.11.17737
Coleman RT, Struhl G (2017) Causal role for inheritance of H3K27me3 in maintaining the off state of a Drosophila HOX gene. Science 356:41. https://doi.org/10.1126/science.aai8236
Copenhaver GP, Pikaard CS (1996) RFLP and physical mapping with an rDNA-specific endonuclease reveals that nucleolus organizer regions of Arabidopsis thaliana adjoin the telomeres on chromosomes 2 and 4. Plant J. https://doi.org/10.1046/j.1365-313X.1996.09020259.x
Dahl TW, Arens SKM (2020) The impacts of land plant evolution on Earth’s climate and oxygenation state—an interdisciplinary review. Chem Geol. https://doi.org/10.1016/j.chemgeo.2020.119665
Dangwal M, Kapoor S, Kapoor M (2014) The PpCMT chromomethylase affects cell growth and interacts with the homolog of LIKE HETEROCHROMATIN PROTEIN 1 in the moss Physcomitrella patens. Plant J. https://doi.org/10.1111/tpj.12406
Derkacheva M, Steinbach Y, Wildhaber T et al (2013) Arabidopsis MSI1 connects LHP1 to PRC2 complexes. EMBO J. https://doi.org/10.1038/emboj.2013.145
Dickinson HG, Grant-Downton R (2009) Bridging the generation gap: flowering plant gametophytes and animal germlines reveal unexpected similarities. Biol Rev Camb Philos Soc 84:589–615. https://doi.org/10.1111/j.1469-185X.2009.00088.x
Dierschke T, Flores-Sandoval E, Rast-Somssich MI et al (2020) Gamete-specific expression of TALE class HD genes activates the diploid sporophyte program in Marchantia polymorpha. bioRxiv
Dimichele WA, Phillips TL (2002) The ecology of Paleozoic ferns. Rev Palaeobot Palynol. https://doi.org/10.1016/S0034-6667(01)00134-8
Domb K, Katz A, Harris KD et al (2021) DNA methylation mutants in Physcomitrella patens elucidate individual roles of CG and non-CG methylation in genome regulation. Proc Natl Acad Sci USA. https://doi.org/10.1073/PNAS.2011361117
Du J, Zhong X, Bernatavichute YV et al (2012) Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell. https://doi.org/10.1016/j.cell.2012.07.034
Du J, Johnson LM, Groth M et al (2014) Mechanism of DNA methylation-directed histone methylation by KRYPTONITE. Mol Cell. https://doi.org/10.1016/j.molcel.2014.06.009
Du J, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. https://doi.org/10.1038/nrm4043
Estève PO, Hang GC, Smallwood A et al (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. https://doi.org/10.1101/gad.1463706
Fan X, Han W, Teng L et al (2020) Single-base methylome profiling of the giant kelp Saccharina japonica reveals significant differences in DNA methylation to microalgae and plants. New Phytol. https://doi.org/10.1111/nph.16125
Feng W, Michaels SD (2015) Accessing the inaccessible: the organization, transcription, replication, and repair of heterochromatin in plants. Annu Rev Genet 49:439–459. https://doi.org/10.1146/annurev-genet-112414-055048
Feng S, Jacobsen SE, Reik W (2010) Epigenetic reprogramming in plant and animal development. Science (new York, NY) 330:622–627. https://doi.org/10.1126/science.1190614
Flores-Sandoval E, Dierschke T, Fisher TJ, Bowman JL (2016) Efficient and inducible use of artificial MicroRNAs in Marchantia polymorpha. Plant Cell Physiol. https://doi.org/10.1093/pcp/pcv068
Frapporti A, Miró Pina C, Arnaiz O et al (2019) The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat Commun. https://doi.org/10.1038/s41467-019-10648-5
Fuchs J, Jovtchev G, Schubert I (2008) The chromosomal distribution of histone methylation marks in gymnosperms differs from that of angiosperms. Chromosome Res. https://doi.org/10.1007/s10577-008-1252-4
Gehring M, Satyaki PR (2017) Endosperm and imprinting, inextricably linked. Plant Physiol 173:143–154. https://doi.org/10.1104/pp.16.01353
Gehring M, Huh JH, Hsieh TF et al (2006) DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell. https://doi.org/10.1016/j.cell.2005.12.034
Gehring M, Bubb KL, Henikoff S (2009) Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science. https://doi.org/10.1126/science.1171609
Gombar S, MacCarthy T, Bergman A (2014) Epigenetics decouples mutational from environmental robustness. Did it also facilitate multicellularity? PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1003450
Graham LKE, Wilcox LW (2000) The origin of alternation of generations in land plants: a focus on matrotrophy and hexose transport. Philosophical Transactions ofthe Royal Society of London. Ser B: Biol Sci 355(1398):757–767. https://doi.org/10.1098/rstb.2000.0614
Grewal SIS, Jia S (2007) Heterochromatin revisited. Nat Rev Genet 8:35–46
Grewal SIS, Klar AJS (1996) Chromosomal inheritance of epigenetic states in fission yeast during mitosis and meiosis. Cell. https://doi.org/10.1016/S0092-8674(00)80080-X
Grossniklaus U, Vielle-Calzada JP, Hoeppner MA, Gagliano WB (1998) Maternal control of embryogenesis by MEDEA, a Polycomb group gene in Arabidopsis. Science 280:446–450. https://doi.org/10.1126/science.280.5362.446
Haig D, Wilczek A (2006) Sexual conflict and the alternation of haploid and diploid generations. Philosophical Transactions of the Royal Society B: Biological Sciences 361(1466):335–343. https://doi.org/10.1098/rstb.2005.1794
Haig D (2008) Homologous versus antithetic alternation of generations and the origin of sporophytes. Bot Rev 74:395–418
Hajkova P (2011) Epigenetic reprogramming in the germline: towards the ground state of the epigenome. Philos Trans R Soc B Biol Sci 366:2266–2273
Hansen KH, Bracken AP, Pasini D et al (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. https://doi.org/10.1038/ncb1787
He S, Vickers M, Zhang J, Feng X (2019) Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. J Res. https://doi.org/10.7554/eLife.42530
Heer K, Ullrich KK, Hiss M et al (2018) Detection of somatic epigenetic variation in Norway spruce via targeted bisulfite sequencing. Ecol Evol. https://doi.org/10.1002/ece3.4374
Heesch S, Serrano-Serrano M, Luthringer R et al (2019) Evolution of life cycles and reproductive traits: insights from the brown algae. bioRxiv. https://doi.org/10.1101/530477
Henikoff S, Ahmad K (2020) Evolution: heterochromatin diversity in early-branching land plants. Curr Biol. https://doi.org/10.1016/j.cub.2019.12.044
Henikoff S, Shilatifard A (2011) Histone modification: cause or cog? Trends Genet 27:389–396
Hennig L, Derkacheva M (2009) Diversity of Polycomb group complexes in plants: same rules, different players? Trends Genet 25:414–423
Hernandez-Lagana E, Autran D (2020) H31 eviction marks female germline precursors in arabidopsis. Plants. https://doi.org/10.3390/plants9101322
Hisanaga T, Yamaoka S, Kawashima T, Higo A, Nakajima K, Araki T, Kohchi T, Berger F (2019) Building newinsights in plant gametogenesis from an evolutionary perspective. Nature Plants 5(7):663–669. https://doi.org/10.1038/s41477-019-0466-0
Hofmeister W (1851) Vergleichende Untersuchungen der Keimung, Entfaltung und Fruchtbildung höherer Kryptogamen (Moose, Farrn, Equisetaceen, Rhizocarpeen und Lycopodiaceen)und der Samenbildung der Coniferen. https://www.worldcat.org/title/vergleichende-untersuchungen-der-keimung-entfaltung-und-fruchtbildunghoherer-kryptogamen-moose-farrn-equisetaceen-rhizocarpeen-und-lycopodiaceen-und-der-samenbildung-derconiferen/oclc/4067212
Hofmeister W (1862) On the germination, development, and fructification of the higher Cryptogamia, and on the fructification of the Coniferae. https://doi.org/10.5962/bhl.title.23191
Holliday R, Pugh JE (1975) DNA modification mechanisms and gene activity during development. Science. https://doi.org/10.1126/science.1111098
Horst NA, Reski R (2016) Alternation of generations—unravelling the underlying molecular mechanism of a 165-year-old botanical observation. Plant Biol (stuttg). https://doi.org/10.1111/plb.12468
Horst NA, Katz A, Pereman I et al (2016) A single homeobox gene triggers phase transition, embryogenesis and asexual reproduction. Nature Plants. https://doi.org/10.1038/nplants.2015.209
Horstman A, Li M, Heidmann I et al (2017) The BABY BOOM transcription factor activates the LEC1-ABI3-FUS3-LEC2 network to induce somatic embryogenesis. Plant Physiol 175:848–857. https://doi.org/10.1104/pp.17.00232
Houben A, Kumke K, Nagaki K, Hause G (2011) CENH3 distribution and differential chromatin modifications during pollen development in rye (Secale cereale L.). Chromosome Res Int J Mol Supramol Evol Asp Chromosome Biol 19:471–480. https://doi.org/10.1007/s10577-011-9207-6
Hsieh TF, Ibarra CA, Silva P et al (2009) Genome-wide demethylation of Arabidopsis endosperm. Science 324:1451–1454. https://doi.org/10.1126/science.1172417
Hu H, Guo X (1999) In vitro induced haploids in plant genetics and breeding. In: Soh WY, Bhojwani SS (eds) Morphogenesis in plant tissue cultures. Springer, Dordrecht, pp 329–361. https://doi.org/10.1007/978-94-015-9253-6_12
Huang F, Zhu QH, Zhu A et al (2017a) Mutants in the imprinted PICKLE RELATED 2 gene suppress seed abortion of fertilization independent seed class mutants and paternal excess interploidy crosses in Arabidopsis. Plant J. https://doi.org/10.1111/tpj.13500
Huang Y, Chen DH, Liu BY et al (2017b) Conservation and diversification of polycomb repressive complex 2 (PRC2) proteins in the green lineage. Brief Funct Genom. https://doi.org/10.1093/bfgp/elw007
Huang Y, Jiang L, Liu BY et al (2019) Evolution and conservation of polycomb repressive complex 1 core components and putative associated factors in the green lineage. BMC Genom. https://doi.org/10.1186/s12864-019-5905-9
Huff JT, Zilberman D (2014) Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell. https://doi.org/10.1016/j.cell.2014.01.029
Ibarra CA, Feng X, Schoft VK et al (2012) Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science (new York, NY) 337:1360–1364. https://doi.org/10.1126/science.1224839
Ikeda Y, Nishihama R, Yamaoka S et al (2018) Loss of CG methylation in Marchantia polymorpha causes disorganization of cell division and reveals unique DNA methylation regulatory mechanisms of non-CG methylation. Plant Cell Physiol 59:2421–2431. https://doi.org/10.1093/pcp/pcy161
Ingouff M, Hamamura Y, Gourgues M et al (2007) Distinct dynamics of HISTONE3 variants between the two fertilization products in plants. Curr Biol CB 17:1032–1037
Ingouff M, Rademacher S, Holec S et al (2010) Zygotic resetting of the HISTONE 3 variant repertoire participates in epigenetic reprogramming in Arabidopsis. Curr Biol CB 20:2137–2143. https://doi.org/10.1016/j.cub.2010.11.012
Jackson JP, Lindroth AM, Cao X, Jacobsen SE (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. https://doi.org/10.1038/nature731
Jacob Y, Feng S, LeBlanc CA et al (2009) ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16:763–768. https://doi.org/10.1038/nsmb.1611
Jacob Y, Bergamin E, Donoghue MTA et al (2014) Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science 343:1249–1253. https://doi.org/10.1126/science.1248357
Jamieson K, Wiles ET, McNaught KJ et al (2016) Loss of HP1 causes depletion of H3K27me3 from facultative heterochromatin and gain of H3K27me2 at constitutive heterochromatin. Genome Res. https://doi.org/10.1101/gr.194555.115
Jarvis EE, Dunahay TG, Brown LM (1992) DNA nucleoside composition and methylation in several species of microalgae. J Phycol. https://doi.org/10.1111/j.0022-3646.1992.00356.x
Jeong BR, Wu-Scharf D, Zhang C, Cerutti H (2002) Suppressors of transcriptional transgenic silencing in Chlamydomonas are sensitive to DNA-damaging agents and reactivate transposable elements. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.022392999
Jiang D, Berger F (2017) DNA replication-coupled histone modification maintains Polycomb gene silencing in plants. Science (new York, NY) 357:1146–1149. https://doi.org/10.1126/science.aan4965
John DM (1994) Alternation of generations in algae: Its complexity maintenance and evolution. Bio Rev 69(3):275–291. https://doi.org/10.1111/j.1469-185X.1994.tb01272.x
Johnsen ST, Junttila O, Dæhlen OG (1996) Influence of the female flowering environment on autumn frost-hardiness of Picea abies progenies. Theor Appl Genet. https://doi.org/10.1007/BF00221890
Johnsen Ø, Fossdal CG, Nagy N et al (2005) Climatic adaptation in Picea abies progenies is affected by the temperature during zygotic embryogenesis and seed maturation. Plant Cell Environ. https://doi.org/10.1111/j.1365-3040.2005.01356.x
Kawashima T, Berger F (2014) Epigenetic reprogramming in plant sexual reproduction. Nat Rev Genet. https://doi.org/10.1038/nrg3685
Khan A, Eikani CK, Khan H et al (2018) Characterization of Chlamydomonas reinhardtii core histones by top-down mass spectrometry reveals unique algae-specific variants and post-translational modifications. J Proteome Res. https://doi.org/10.1021/acs.jproteome.7b00780
Khanday I, Skinner D, Yang B et al (2019) A male-expressed rice embryogenic trigger redirected for asexual propagation through seeds. Nature 565:91–95. https://doi.org/10.1038/s41586-018-0785-8
Khouider S, Borges F, LeBlanc C et al (2021) Male fertility in Arabidopsis requires active DNA demethylation of genes that control pollen tube function. Nat Commun. https://doi.org/10.1038/s41467-020-20606-1
Kiyosue T, Ohad N, Yadegari R et al (1999) Control of fertilization-independent endosperm development by the MEDEA polycomb gene in Arabidopsis. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.96.7.4186
Köhler C, Makarevich G (2006) Epigenetic mechanisms governing seed development in plants. EMBO Rep 7:1223–1227
Köhler C, Hennig L, Bouveret R et al (2003) Arabidopsis MSI1 is a component of the MEA/FIE Polycomb group complex and required for seed development. EMBO J. https://doi.org/10.1093/emboj/cdg444
Köhler C, Mittelsten Scheid O, Erilova A (2010) The impact of the triploid block on the origin and evolution of polyploid plants. Trends Genet 26:142–148
Kradolfer D, Wolff P, Jiang H et al (2013) An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev Cell 26:525–535. https://doi.org/10.1016/j.devcel.2013.08.006
Kronholm I, Bassett A, Baulcombe D, Collins S (2017) Epigenetic and genetic contributions to adaptation in chlamydomonas. Mol Biol Evol. https://doi.org/10.1093/molbev/msx166
Kumar S, Stecher G, Suleski M, Hedges SB (2017) TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. https://doi.org/10.1093/molbev/msx116
Lafon-Placette C, Johannessen IM, Hornslien KS et al (2017) Endosperm-based hybridization barriers explain the pattern of gene flow between Arabidopsis lyrata and Arabidopsis arenosa in Central Europe. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1615123114
Laprell F, Finkl K, Müller J (2017) Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science (new York, NY) 356:85–88. https://doi.org/10.1126/science.aai8266
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220
Lee JH, Lin H, Joo S, Goodenough U (2008) Early sexual origins of homeoprotein heterodimerization and evolution of the plant KNOX/BELL family. Cell. https://doi.org/10.1016/j.cell.2008.04.028
Lee JM, Yang EC, Graf L et al (2018) Analysis of the draft genome of the red seaweed gracilariopsis chorda provides insights into genome size evolution in rhodophyta. Mol Biol Evol. https://doi.org/10.1093/molbev/msy081
Lenormand T, Engelstädter J, Johnston SE et al (2016) Evolutionary mysteries in meiosis. Philos Trans R Soc B Biol Sci 371:20160001
Li F, Martienssen R, Cande WZ (2011) Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature. https://doi.org/10.1038/nature10161
Li Z, Fu X, Wang Y et al (2018) Polycomb-mediated gene silencing by the BAH–EMF1 complex in plants. Nat Genet. https://doi.org/10.1038/s41588-018-0190-0
Lindroth AM, Cao X, Jackson JP et al (2001) Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. https://doi.org/10.1126/science.1059745
Lopez D, Hamaji T, Kropat J et al (2015) Dynamic changes in the transcriptome and methylome of Chlamydomonas reinhardtii throughout its life cycle. Plant Physiol. https://doi.org/10.1104/pp.15.00861
Loppin B, Bonnefoy E, Anselme C et al (2005) The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 437:1386–1390. https://doi.org/10.1038/nature04059
Loyola A, Tagami H, Bonaldi T et al (2009) The HP1α-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. https://doi.org/10.1038/embor.2009.90
Luo M, Bilodeau P, Koltunow A et al (1999) Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.96.1.296
Luo X, Ou Y, Li R, He Y (2020) Maternal transmission of the epigenetic ‘memory of winter cold’ in Arabidopsis. Nat Plants. https://doi.org/10.1038/s41477-020-00774-0
Lv J, Yu K, Wei J et al (2020) Generation of paternal haploids in wheat by genome editing of the centromeric histone CENH3. Nat Biotechnol. https://doi.org/10.1038/s41587-020-0728-4
Malik G, Dangwal M, Kapoor S, Kapoor M (2012) Role of DNA methylation in growth and differentiation in Physcomitrella patens and characterization of cytosine DNA methyltransferases. FEBS J. https://doi.org/10.1111/febs.12002
Margueron R, Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature 469:343–349. https://doi.org/10.1038/nature09784
Margueron R, Justin N, Ohno K et al (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. https://doi.org/10.1038/nature08398
Matyášek R, Krumpolcová A, Lunerová J et al (2019) Unique epigenetic features of ribosomal RNA genes (rDNA) in early diverging plants (bryophytes). Front Plant Sci. https://doi.org/10.3389/fpls.2019.01066
Matzke M, Kanno T, Daxinger L et al (2009) RNA-mediated chromatin-based silencing in plants. Curr Opin Cell Biol 21:367–376
Maumus F, Rabinowicz P, Bowler C, Rivarola M (2011) Stemming epigenetics in marine stramenopiles. Curr Genom. https://doi.org/10.2174/138920211796429727
Mérai Z, Chumak N, García-Aguilar M et al (2014) The AAA-ATPase molecular chaperone Cdc48/p97 disassembles sumoylated centromeres, decondenses heterochromatin, and activates ribosomal RNA genes. Proc Natl Acad Sci USA 111:16166–16171. https://doi.org/10.1073/pnas.1418564111
Mikulski P, Komarynets O, Fachinelli F et al (2017) Characterization of the polycomb-group mark H3K27me3 in unicellular algae. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00607
Montgomery SA, Tanizawa Y, Galik B et al (2020) Chromatin organization in early land plants reveals an ancestral association between H3K27me3, transposons, and constitutive heterochromatin. Curr Biol. https://doi.org/10.1016/j.cub.2019.12.015
Moreno-Romero J, del Toro-De LG, Yadav VK et al (2019) Epigenetic signatures associated with imprinted paternally expressed genes in the Arabidopsis endosperm. Genome Biol 20:41. https://doi.org/10.1186/s13059-019-1652-0
Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Genet 14suppl_1:R47–R58. https://doi.org/10.1007/s00497-021-00422-314
Mosquna A, Katz A, Decker EL et al (2009) Regulation of stem cell maintenance by the Polycomb protein FIE has been conserved during land plant evolution. Development 136:2433–2444. https://doi.org/10.1242/dev.035048
Müller DG (1966) Untersuchungen zur Entwicklungsgeschichte der Braunalge Ectocarpus siliculosus Aus Neapel. Planta. https://doi.org/10.1007/BF00385371
Nakamura M, Köhler C, Hennig L (2019) Tissue-specific transposon-associated small RNAs in the gymnosperm tree, Norway spruce. BMC Genom. https://doi.org/10.1186/s12864-019-6385-7
Nakamura M, Batista RA, Köhler C, Hennig L (2020) Polycomb Repressive Complex 2-mediated histone modification H3K27me3 is associated with embryogenic potential in Norway spruce. J Exp Bot. https://doi.org/10.1093/jxb/eraa365
Nakayama J, Rice JC, Strahl BD et al (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. https://doi.org/10.1126/science.1060118
Neupert J, Gallaher SD, Lu Y et al (2020) An epigenetic gene silencing pathway selectively acting on transgenic DNA in the green alga Chlamydomonas. Nat Commun. https://doi.org/10.1038/s41467-020-19983-4
Niklas KJ, Cobb ED, Kutschera U (2014) Did meiosis evolve before sex and the evolution of eukaryotic life cycles? BioEssays. https://doi.org/10.1002/bies.201400045
Noy-Malka C, Yaari R, Itzhaki R et al (2014) A single CMT methyltransferase homolog is involved in CHG DNA methylation and development of Physcomitrella patens. Plant Mol Biol. https://doi.org/10.1007/s11103-013-0165-6
O’Brown ZK, Boulias K, Wang J et al (2019) Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genom. https://doi.org/10.1186/s12864-019-5754-6
O’Malley RC, Huang SSC, Song L et al (2016) Cistrome and epicistrome features shape the regulatory DNA landscape. Cell. https://doi.org/10.1016/j.cell.2016.04.038
Ohad N, Margossian L, Hsu YC et al (1996) A mutation that allows endosperm development without fertilization. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.93.11.5319
Ohad N, Yadegari R, Margossian L et al (1999) Mutations in FIE, a WD polycomb group gene, allow endosperm development without fertilization. Plant Cell 11:407–415. https://doi.org/10.1105/tpc.11.3.407
Okano Y, Aono N, Hiwatashi Y et al (2009) A polycomb repressive complex 2 gene regulates apogamy and gives evolutionary insights into early land plant evolution. Proc Natl Acad Sci USA 106:16321–16326. https://doi.org/10.1073/pnas.0906997106
Park K, Kim MY, Vickers M et al (2016) DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1619047114
Park JS, Frost JM, Park K et al (2017) Control of DEMETER DNA demethylase gene transcription in male and female gamete companion cells in Arabidopsis thaliana. Proc Natl Acad Sci USA 114:2078–2083. https://doi.org/10.1073/pnas.1620592114
Pei L, Zhang L, Li J et al (2019) Tracing the origin and evolution history of methylation-related genes in plants. BMC Plant Biol. https://doi.org/10.1186/s12870-019-1923-7
Pereman I, Mosquna A, Katz A et al (2016) The Polycomb group protein CLF emerges as a specific tri-methylase of H3K27 regulating gene expression and development in Physcomitrella patens. Biochim Biophys Acta Gene Regul Mech. https://doi.org/10.1016/j.bbagrm.2016.05.004
Pérez-Lluch S, Blanco E, Tilgner H et al (2015) Absence of canonical marks of active chromatin in developmentally regulated genes. Nat Genet 47:1158–1167. https://doi.org/10.1038/ng.3381
Petryk N, Dalby M, Wenger A et al (2018) MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science. https://doi.org/10.1126/science.aau0294
Pillot M, Baroux C, Vazquez MA et al (2010) Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell. https://doi.org/10.1105/tpc.109.071647
Puttick MN, Morris JL, Williams TA et al (2018) The interrelationships of land plants and the nature of the ancestral embryophyte. Curr Biol. https://doi.org/10.1016/j.cub.2018.01.063
Rabanal FA, Mandáková T, Soto-Jiménez LM et al (2017) Epistatic and allelic interactions control expression of ribosomal RNA gene clusters in Arabidopsis thaliana. Genome Biol. https://doi.org/10.1186/s13059-017-1209-z
Rebollo R, Romanish MT, Mager DL (2012) Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet. https://doi.org/10.1146/annurev-genet-110711-155621
Reddington JP, Perricone SM, Nestor CE et al (2013) Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. https://doi.org/10.1186/gb-2013-14-3-r25
Reese BE, Bachman KE, Baylin SB, Rountree MR (2003) The methyl-CpG binding protein MBD1 interacts with the p150 subunit of chromatin assembly factor 1. Mol Cell Biol. https://doi.org/10.1128/mcb.23.9.3226-3236.2003
Reinberg D, Vales LD (2018) Chromatin domains rich in inheritance. Science 361:33–34. https://doi.org/10.1126/science.aat7871
Rensing SA, Lang D, Zimmer AD et al (2008) The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. https://doi.org/10.1126/science.1150646
Reverón-Gómez N, González-Aguilera C, Stewart-Morgan KR et al (2018) Accurate recycling of parental histones reproduces the histone modification landscape during DNA replication. Mol Cell 72:239-249.e5. https://doi.org/10.1016/j.molcel.2018.08.010
Reynolds WF, Wolfe SL (1978) Changes in basic proteins during sperm maturation in a plant, Marchantia polymorpha. Exp Cell Res 116:269–273. https://doi.org/10.1016/0014-4827(78)90448-2
Rougée M, Quadrana L, Zervudacki J et al (2019) Altering PRC2 activity partially suppresses ddm1 mutant phenotypes in Arabidopsis. bioRxiv. https://doi.org/10.1101/782219
Rudall PJ (2006a) How many nuclei make an embryo sac in flowering plants? BioEssays 28:1067–1071
Rudall PJ (2006b) How many nuclei make an embryo sac in flowering plants? BioEssays 28:1067–1071
Rudall PJ, Bateman RM (2007a) Developmental bases for key innovations in the seed-plant microgametophyte. Trends Plant Sci 12:317–326. https://doi.org/10.1016/j.tplants.2007.06.004
Sakakibara K, Ando S, Yip HK et al (2013) KNOX2 genes regulate the haploid-to-diploid morphological transition in land plants. Science. https://doi.org/10.1126/science.1230082
Sano Y, Tanaka I (2010) Distinct localization of histone H3 methylation in the vegetative nucleus of lily pollen. Cell Biol Int 34:253–259. https://doi.org/10.1042/CBI20090124
Sarraf SA, Stancheva I (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 15:595–605
Schlissel G, Rine J (2019) The nucleosome core particle remembers its position through DNA replication and RNA transcription. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1911943116
Schmid MW, Giraldo-Fonseca A, Rövekamp M et al (2018) Extensive epigenetic reprogramming during the life cycle of Marchantia polymorpha. Genome Biol 19:9. https://doi.org/10.1186/s13059-017-1383-z
Schoft VK, Chumak N, Mosiolek M et al (2009) Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep 10:1015–1021. https://doi.org/10.1038/embor.2009.152
Schoft VK, Chumak N, Choi Y et al (2011) Function of the DEMETER DNA glycosylase in the Arabidopsis thaliana male gametophyte. Proc Natl Acad Sci USA 108:8042–8047. https://doi.org/10.1073/pnas.1105117108
Schubert D (2019) Evolution of Polycomb-group function in the green lineage. F1000REsearch. https://doi.org/10.12688/f1000research.16986.1
Schwartz YB, Pirrotta V (2007) Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 8:9–22
Searles RB (1980) The strategy of the red algal life history. Am Nat. https://doi.org/10.1086/283548
Shaver S, Casas-Mollano JA, Cerny RL, Cerutti H (2010) Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics. https://doi.org/10.4161/epi.5.4.11608
She W, Baroux C (2015) Chromatin dynamics in pollen mother cells underpin a common scenario at the somatic-to-reproductive fate transition of both the male and female lineages in Arabidopsis. Front Plant Sci 6:294. https://doi.org/10.3389/fpls.2015.00294
She W, Grimanelli D, Rutowicz K et al (2013) Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development (cambridge, England) 140:4008–4019. https://doi.org/10.1242/dev.095034
Shih PM, Matzke NJ (2013) Primary endosymbiosis events date to the later Proterozoic with cross-calibrated phylogenetic dating of duplicated ATPase proteins. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1305813110
Slotkin RK, Vaughn M, Borges F et al (2009) Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136:461–472
Stewart KD, Mattox KR (1975) Comparative cytology, evolution and classification of the green algae with some consideration of the origin of other organisms with chlorophylls A and B. Bot Rev. https://doi.org/10.1007/BF02860837
Stroud H, Do T, Du J et al (2014) Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol. https://doi.org/10.1038/nsmb.2735
Sun B, Looi LS, Guo S et al (2014) Timing mechanism dependent on cell division is invoked by Polycomb eviction in plant stem cells. Science 343:1248559–1248559. https://doi.org/10.1126/science.1248559
Tonosaki K, Sekine D, Ohnishi T et al (2018) Overcoming the species hybridization barrier by ploidy manipulation in the genus Oryza. Plant J. https://doi.org/10.1111/tpj.13803
Torres-Garcia S, Yaseen I, Shukla M et al (2020) Epigenetic gene silencing by heterochromatin primes fungal resistance. Nature. https://doi.org/10.1038/s41586-020-2706-x
Traller JC, Cokus SJ, Lopez DA et al (2016) Genome and methylome of the oleaginous diatom Cyclotella cryptica reveal genetic flexibility toward a high lipid phenotype. Biotechnol Biofuels. https://doi.org/10.1186/s13068-016-0670-3
Tsuboi H, Sutoh K, Wada M (2012) Epigenetic memory of DNAi associated with cytosine methylation and histone modification in fern. Plant Signal Behav. https://doi.org/10.4161/psb.21974
Turck F, Roudier F, Farrona S et al (2007) Arabidopsis TFL2/LHP1 specifically associates with genes marked by trimethylation of histone H3 lysine 27. PLoS Genet. https://doi.org/10.1371/journal.pgen.0030086
van Dijk K, Marley KE, Jeong BR et al (2005) Monomethyl histone H3 lysine 4 as an epigenetic mark for silenced euchromatin in Chlamydomonas. Plant Cell. https://doi.org/10.1105/tpc.105.034165
Veluchamy A, Lin X, Maumus F et al (2013) Insights into the role of DNA methylation in diatoms by genome-wide profiling in Phaeodactylum tricornutum. Nat Commun. https://doi.org/10.1038/ncomms3091
Veluchamy A, Rastogi A, Lin X et al (2015) An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol. https://doi.org/10.1186/s13059-015-0671-8
Walker J, Gao H, Zhang J et al (2018) Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat Genet. https://doi.org/10.1038/s41588-017-0008-5
Walker J, Zhang J, Liu Y et al (2021) Extensive N4 cytosine methylation is essential for marchantia sperm function. bioRxiv. https://doi.org/10.1101/2021.02.12.428880
Wang G, Jiang H, del TorodeLeón G et al (2018) Sequestration of a transposon-derived siRNA by a target mimic imprinted gene induces postzygotic reproductive isolation in Arabidopsis. Dev Cell 46:10. https://doi.org/10.1016/j.devcel.2018.07.014
Wang Z, Butel N, Santos-González J et al (2020) Polymerase IV plays a crucial role in pollen development in Capsella. Plant Cell. https://doi.org/10.1105/tpc.19.00938
Wang N, Gent JI, Dawe RK (2021) Haploid induction by a maize cenh3 null mutant. Sci Adv. https://doi.org/10.1126/sciadv.abe2299
Warne TR, Hickok LG (1991) Control of sexual development in gametophytes of Ceratopteris richardii: antheridiogen and abscisic acid. Bot Gaz. https://doi.org/10.1086/337874
Weinhofer I, Hehenberger E, Roszak P et al (2010) H3K27me3 profiling of the endosperm implies exclusion of polycomb group protein targeting by DNA methylation. PLoS Genet. https://doi.org/10.1371/journal.pgen.1001152
Wibowo A, Becker C, Marconi G et al (2016) Hyperosmotic stress memory in arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by dna glycosylase activity. Elife. https://doi.org/10.7554/eLife.13546
Widiez T, Symeonidi A, Luo C et al (2014) The chromatin landscape of the moss Physcomitrella patens and its dynamics during development and drought stress. Plant J. https://doi.org/10.1111/tpj.12542
Wiles ET, McNaught KJ, Kaur G et al (2020) Evolutionarily ancient BAH-PHD protein mediates Polycomb silencing. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1918776117
Wilkins AS, Holliday R (2009) The evolution of meiosis from mitosis. Genetics 181:3–12
Wolff P, Jiang H, Wang G et al (2015) Paternally expressed imprinted genes establish postzygotic hybridization barriers in Arabidopsis thaliana. Elife. https://doi.org/10.7554/eLife.10074
Woo HR, Dittmer TA, Richards EJ (2008) Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in arabidopsis. PLoS Genet. https://doi.org/10.1371/journal.pgen.1000156
Yaari R, Noy-Malka C, Wiedemann G et al (2015) DNA METHYLTRANSFERASE 1 is involved in mCG and mCCG DNA methylation and is essential for sporophyte development in Physcomitrella patens. Plant Mol Biol 88:387–400. https://doi.org/10.1007/s11103-015-0328-8
Yaari R, Katz A, Domb K et al (2019) RdDM-independent de novo and heterochromatin DNA methylation by plant CMT and DNMT3 orthologs. Nat Commun. https://doi.org/10.1038/s41467-019-09496-0
Yakovlev I, Fossdal CG, Skrøppa T et al (2012) An adaptive epigenetic memory in conifers with important implications for seed production. Seed Sci Res 22:63–76
Yang Z, Qian S, Scheid RN et al (2018) EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat Genet 50:1247–1253
Ye P, Luan Y, Chen K et al (2017) MethSMRT: an integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw950
Zemach A, Zilberman D (2010) Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol 20:R780–R785
Zemach A, Kim MY, Hsieh PH et al (2013) The arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. https://doi.org/10.1016/j.cell.2013.02.033
Zhang X, Germann S, Blus BJ et al (2007) The Arabidopsis LHP1 protein colocalizes with histone H3 Lys27 trimethylation. Nat Struct Mol Biol. https://doi.org/10.1038/nsmb1283
Zhang H, Luo M, Johnson SD et al (2016) Parental genome imbalance causes post-zygotic seed lethality and deregulates imprinting in rice. Rice. https://doi.org/10.1186/s12284-016-0115-4
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19:489–506
Zhao X, Rastogi A, Deton Cabanillas AF et al (2021) Genome wide natural variation of H3K27me3 selectively marks genes predicted to be important for cell differentiation in Phaeodactylum tricornutum. New Phytol. https://doi.org/10.1111/nph.17129
Zheng H, Huang B, Zhang B et al (2016) Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63:1066–1079. https://doi.org/10.1016/j.molcel.2016.08.032
Zhong B, Sun L, Penny D (2015) The origin of land plants: a phylogenomic perspective. Evol Bioinf. https://doi.org/10.4137/EBO.S29089
Zhu B, Reinberg D (2011) Epigenetic inheritance: uncontested? Cell Res 21:435–441
Acknowledgements
We thank Frederic Berger for his insight and discussions that helped inspire concepts discussed in this review. We would like to thank Susana Coelho for critical reading and discussion on sections relating to brown algae. The authors were financially supported by the Max Planck Society.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
JV contributed to the research and sections relating to diatoms and algae. MB conceived the review, prepared figures and wrote the manuscript. Both authors read and approved the manuscript.
Corresponding author
Additional information
Communicated by Frederic Berger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article belongs to special issue S.I.: Evolution of plant reproduction
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vigneau, J., Borg, M. The epigenetic origin of life history transitions in plants and algae. Plant Reprod 34, 267–285 (2021). https://doi.org/10.1007/s00497-021-00422-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00497-021-00422-3