
Abstract
Key message
We report the adaptation of the INTACT method for RNA-sequencing in the endosperm and demonstrate its feasibility for allele-specific expression analysis.
Abstract
Tissue-specific transcriptome analyses provide important insights into the developmental programs of defined cell types. The isolation of nuclei tagged in specific cell types (INTACT) is a versatile method that allows to isolate highly pure nuclei from defined tissue types that can be used for several downstream applications. Here, we describe the adaptation of INTACT from endosperm nuclei for high-throughput RNA-sequencing. By analyzing the ratio of parental reads and tissue-specific gene expression in the endosperm, we could assess the contamination level of our samples. Based on this analysis, we estimate that in most of the samples the contamination level is lower than in previously published datasets. We further show that the nuclear transcriptome and total transcriptome of the endosperm are well correlated. Together, our data show that INTACT of the endosperm is a reliable methodology for endosperm-specific transcriptome analysis that overcomes the limitation of time-consuming manual endosperm dissection that is connected with high levels of maternal tissue contamination. INTACT does not rely on expensive equipment and can be set up in every standard molecular biology laboratory, making it the method of choice for future molecular studies of the endosperm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Double fertilization in flowering plants results in the formation of the embryo and the endosperm, a nurturing tissue similar to the placenta in mammals that supports embryo development (Yan et al. 2014). The endosperm is surrounded by multiple maternal sporophytic cell layers, hindering its isolation for cell-specific analyses. In particular, for species forming small seeds such as Arabidopsis thaliana the isolation of endosperm in sufficient quality and quantity is a substantial challenge. So far, the majority of Arabidopsis endosperm transcriptome data were generated from manually dissected endosperm and were found to have substantial levels of maternal tissue contamination (Gehring et al. 2011; Hsieh et al. 2011; Pignatta et al. 2014; Schon and Nodine 2017). Alternative techniques such as fluorescence-activated cell sorting (FACS) and laser capture microdissection (LCM) have also been employed for the generation of transcriptome and epigenome profiles of the Arabidopsis endosperm (Le et al. 2010; Weinhofer et al. 2010); however, the requirement of large amounts of starting material and expensive flow cytometry facilities limit the general application of these techniques. For this reason, there is the need to develop versatile, inexpensive and highly reproducible techniques to facilitate genome-wide studies of the endosperm. INTACT technique allows rapid and efficient nuclei isolation from specific cell types without the need for specialized and expensive equipment (Deal and Henikoff 2011; Moreno-Romero et al. 2017). The methodology is based on biotin tagging of cell-specific nuclei, allowing their subsequent purification from the total nuclei pool using streptavidin-coated magnetic beads. The INTACT system requires the co-expression of two components; the first is a synthetic nuclear targeting fusion (NTF) protein composed of the nuclear envelope WPP domain of the Arabidopsis RAN GTPase activating protein 1 (RanGAP1) attached to GFP and to a biotin ligase recognition peptide, which functions as the substrate for the second component, the Escherichia coli biotin ligase (BirA) (Deal and Henikoff 2011).
INTACT was originally developed for the purification of nuclei from Arabidopsis root tissues, but it has been adapted and employed for genomic, epigenomic and proteomic studies of other cell types and plant species such as rice or tomato, as well as non-plant model organisms such as flies, worms and Xenopus (Deal and Henikoff 2010; Steiner et al. 2012; Henry et al. 2012; Amin et al. 2014; Moreno-Romero et al. 2016; Reynoso et al. 2017; Palovaara et al. 2017). Previously, we have reported the adaptation of the INTACT system for the generation of epigenome profiles of the endosperm by expressing the NTF-GFP and BirA components under the endosperm-specific PHERES1 (PHE1) promoter (Moreno-Romero et al. 2016, 2017). PHE1 is imprinted and specifically paternally expressed (Köhler et al. 2005); however, since imprinted expression of PHE1 requires regulatory elements outside of the promoter region (Villar et al. 2009), the INTACT lines are not imprinted. Here, we describe the application of INTACT for RNA-sequencing (RNA-seq) of endosperm-specific nuclei, demonstrating its feasibility and high potential for transcriptome studies. Furthermore, as proof of concept we show the applicability of endosperm INTACT for the generation of parent-of-origin-specific expression data.
Materials and methods
Plant material and growth conditions
We made use of the pistillata-1 (pi-1) mutant (Goto and Meyerowitz 1994) (NASC stock number NW77) and the standard reference Col-0 (NASC stock number N22625). Transgenic Arabidopsis lines expressing PHE1::NTF and PHE1::BirA (lines referred as INT hereafter) are in the Col-0 accession in both wild-type (NASC stock number N2107349) and the delayed dehiscence 2 (dde2) (Przybyla et al. 2008) mutant background (NASC stock number N2107350). Seeds were surface-sterilized by incubating them for 10 min in 70% ethanol and washing them three times with sterile water. Sterilized seeds were sown on MS plates containing 0.5% sucrose and 0.8% agar, stratified for 2–3 days at 4 °C and germinated under long-day conditions (16 h light/8 h darkness) at 21 °C. Seedlings were transferred to soil after 10–12 days and incubated in growth rooms under long-day conditions.
Crosses and starting material
For parent-of-origin transcriptome analyses, we performed Col × Ler (Landsberg erecta) reciprocal crosses as described previously in Moreno-Romero et al. (2017). To facilitate the crosses, we used the male sterile pi-1 (in the Ler accession) and the INT line in the male sterile dde2 background (in the Col accession) as the female parents and pollinated them with the INT line (Col accession) and Ler wild type, respectively. Siliques were collected at 4 days after pollination (DAP). Samples of 250 mg were wrapped in aluminum foil and quickly frozen in liquid nitrogen. Three independent biological replicates for each cross direction were generated.
Nuclei isolation and microscopy
Tissue homogenization was performed as described previously in Moreno-Romero et al. (2017). The samples were incubated under rotation for 30 min with 18 µl of pre-blocked streptavidin dynabeads. Before use, the M-280 streptavidin dynabeads were washed twice with PBSB buffer and pre-incubated on a rotator for 1 h with PBSB. After sample incubation, the beads were collected with a magnet rack and resuspended in 500 µl of PBSBt (PBSB with 0.1% Triton). Finally, the samples were pooled in a Falcon tube containing 11 ml of PBSBt and incubated for 15 min under rotation. The beads were collected using a magnet rack and resuspended in 400 µl of RNAlater (Sigma-Aldrich). At this step, GFP-positive nuclei were identified using a Leica DMI4000B Florescence Microscope equipped with an L5 filter for GFP.
RNA extraction and library preparation
Total RNA was extracted from the resuspended nuclei using the mirVana Isolation Kit Protocol (Ambion) according to the manufacturer’s instructions omitting the enrichment procedure for small RNAs. Subsequent mRNA extraction was performed using NEBNext Poly(A) mRNA Magnetic Isolation, and the Libraries were prepared with the NEBNext Ultra II RNA Library Prep Kit for Illumina and sequenced at the National Genomic Infrastructure (NGI) from SciLife Laboratory (Uppsala, Sweden) on an Illumina HiSeq 2500 in paired-end 125 bp read length.
mRNA-sequencing data processing
Reads were trimmed by removing 15 bp from the 5′ end and mapped in single-end mode to the Arabidopsis (TAIR10) genome previously masked for rRNA genes and for the SNP positions between the TAIR10 (Col) and Ler genome using TopHat v2.1 (Trapnell et al. 2009) (parameters adjusted as -g 1 -a 10 -i 40 -I 5000 -F 0 -r 130). Gene expression was normalized to reads per kilobase per million mapped reads (RPKM) using GFOLD40 (Feng et al. 2012). To discriminate between maternal and paternal transcripts, reads were assigned to the Col or Ler genomes using single nucleotide polymorphisms (SNPs) between the strains. SNP calling was performed with SNPsplit v0.2.0 sorting the mapped reads by parent (Krueger and Andrews 2016).
Results and discussion
Nuclei isolation and contamination assessment
Here, we describe the use of the INTACT method for transcriptome studies by RNA-seq and parent-of-origin-specific analyses. The original data this protocol is based on have been recently published (Moreno-Romero et al. 2018, bioRxiv). We generated parental-specific transcriptome profiles of the endosperm by reciprocally crossing of Col × Ler accessions using endosperm-specific INT lines (Fig. 1). We collected siliques at 4 DAP and followed previously established procedures for tissue homogenization and nuclei purification (Moreno-Romero et al. 2017). After nuclei isolation, we observed abundant GFP-positive nuclei under the fluorescence microscope that were bound by streptavidin dynabeads (Fig. 2). Since purity estimates based on the relative number of NTF-positive and NTF-negative nuclei highly underestimates the contamination level (Moreno-Romero et al. 2017), we estimated sample purity based on the ratio of parental reads obtained after sequencing, following previously established formula (Moreno-Romero et al. 2017). The contamination of our samples ranged between 0 and 15% (Moreno-Romero et al. 2018, bioRxiv), which is comparable to previous experiments (3–11% in Moreno-Romero et al. 2017). We noted that the amount of starting material affects the level of maternal tissue contamination (Table S1); in samples where we used 500 mg of silique material, the contamination level was higher compared to those samples where only 250 mg was used (39% contamination compared to 9–15% in Col × Ler crosses; 10% contamination compared to 0–4% in Ler × Col crosses). We furthermore assessed contamination from non-endosperm tissues by testing for enrichment of tissue-specific transcripts using the pipeline published by Schon and Nodine (2017). Since our samples correspond to endosperm at 4 DAP with developing seeds containing late globular to early heart stage embryos, we compared our dataset with published datasets at the corresponding developmental stages. Low levels of non-endosperm tissues were detected in all samples except in the 500 mg Col × Ler replicate 1 supporting our previous observation of higher contamination in this particular sample. Together with the SNP-based estimate of contamination levels, we conclude that in the 250 mg replicates the contamination levels are low, demonstrating that INTACT is a reliable method for endosperm-specific transcriptomic analysis. In contrast to manually dissected endosperm, we observed a chalazal endosperm tissue enrichment in all our samples (Fig. 3). Although our INT line is expressed in all endosperm compartments (Moreno-Romero et al. 2016), it is more strongly expressed in the chalazal region, which may cause a preferential purification of chalazal endosperm nuclei. Alternatively, the manually dissected endosperm is depleted of the chalazal endosperm that due to its tight association with the seed coat may be less easily captured by manual dissection. Based on the current data, it is not possible to distinguish between both possibilities. As recently reported by Schon and Nodine (2017), available transcriptome datasets of the endosperm contain substantial fractions of seed coat transcripts. From 12 endosperm datasets obtained by manual dissection, 11 show contamination levels above 5%. In contrast, from our 6 INTACT libraries only one shows more than 5% of contamination (Fig. 3). Our data thus demonstrate that the INTACT method outperforms manual dissection of the endosperm in terms of tissue purity, and allows endosperm-specific transcript enrichment comparable to samples obtained by LCM. Additionally, INTACT allows access to earlier developmental stages without compromising the purity of the samples (Fig. 3).

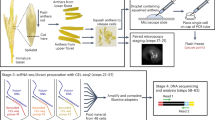
Nuclear transcriptome workflow applying INTACT. INTACT protocol (green), RNA extraction library preparation and sequence treatment (yellow), contamination assessment (red) and allele-specific expression analyses (purple)

Nuclei isolation using INT lines for endosperm. a Examples of NTF-positive nuclei (white arrows) after INTACT and before RNA extraction. b Magnification of a GFP-positive nucleus bound to streptavidin-coated magnetic beads. Bars = 10 μm

Tissue enrichment analysis. Comparison of RNA-seq datasets with our INTACT RNA-seq using the pipeline by Schon and Nodine (2017). We used published datasets of manually dissected endosperm transcriptomes and our datasets generated using INTACT to compare the contamination levels obtained with both methodologies. We compared our 4 DAP endosperm datasets to datasets of samples generated from seeds containing globular and heart stage embryos. *Sample obtained by LCM. NA not available
Previous work established that the nuclear and total cellular mRNA pools are generally comparable (Barthelson et al. 2007; Jacob et al. 2007; Deal and Henikoff 2010). However, since the endosperm is a triploid tissue and thus differs from other vegetative tissues, we tested whether nuclear and cellular mRNAs also correlate in the endosperm. We analyzed the correlation of our nuclear endosperm-specific RNA-seq expression data with previously published manually dissected endosperm of 6 DAP Col × Ler and Ler × Col seed transcriptomes (Pignatta et al. 2014). With the exception of replicates 1 of both cross directions, the correlation values for all other samples were high (Spearman correlation = 0.76 on average) (Supplementary Fig. 1), revealing that nuclear mRNA transcriptomes of the endosperm correlate with total mRNA transcriptomes. Therefore, INTACT is a suitable technique to generate endosperm-specific transcriptome profiles.
Parent-of-origin-specific gene expression in the endosperm
Genomic imprinting is an epigenetic phenomenon causing parental alleles to be differentially expressed. In plants, genomic imprinting is mainly confined to the endosperm and unlike animals, shows high variation among species but also among accessions (Hatorangan et al. 2016; Pignatta et al. 2014). Although inconsistencies among studies are partly explained by natural variation of genomic imprinting, maternal tissue contamination has hindered the accurate estimation of maternally and paternally expressed genes (Pignatta et al. 2014; Schon and Nodine 2017). In addition, lack of formal consensus criteria to call maternally expressed genes (MEGs) and paternally expressed genes (PEGs) in the literature has contributed to the discrepancies in parent-of-origin expression analyses. Here, we describe an easily applicable pipeline to identify imprinted genes in INTACT-generated RNA-seq samples that is based on a combination of statistical thresholds and defined allelic ratios, as previously described (Pignatta et al. 2014). We furthermore define criteria to identify non-canonical allelically biased genes (Fig. 4a; Table 1). We apply this pipeline to our INTACT-purified endosperm RNA-seq data (Moreno-Romero et al. 2018, bioRxiv) and to publicly available RNA-seq data for Col × Ler 6 DAP endosperm to compare our observations (Pignatta et al. 2014).

Parentally biased expression in Col × Ler hybrid endosperm. a Overview of the Col × Ler allele-specific expression analysis. The graph is showing the maternal/total reads values for the reciprocal crosses. Categories are as indicated in Table 1. b Comparison of previously reported MEGs and PEGs (Wolff et al. 2011; Pignatta et al. 2014; Gehring et al. 2011; Hsieh et al. 2011) with our INTACT RNA-seq data. c Comparison of the accession-specific imprinted genes reported by Pignatta et al. (2014) with those identified in this study. d Comparison of accession-specific non-imprinted genes reported by Pignatta et al. (2014) with those identified in this study. Statistical significance of the overlap was calculated using a hypergeometric test (*P < 0.001). PEGs paternally expressed genes, poPEGs potential paternally expressed genes, MEGs maternally expressed genes, poMEGs potential maternally expressed genes, asMEGs accession-specific maternally expressed genes, asPEGs accession-specific paternally expressed genes. Direction of the cross for asMEGs and asPEGs is indicated
Based on our contamination analysis (Fig. 3) and correlation with total cellular transcriptomes (Supplementary Fig. 1), we discarded replicates 1 for both directions of the crosses and merged libraries from two replicates of Col × Ler and Ler × Col for downstream analyses. To ensure statistical significance of the analyses, we defined a minimum of 20 informative reads per analyzed genotype. Statistically significant deviations between maternal and paternal read counts for each gene were tested using Pearson’s Chi-square test, with P values being corrected using the Benjamini–Hochberg method. We furthermore defined allelic thresholds, requiring MEGs to have at least 85% of informative reads derived from the maternal allele, while PEGs to have a minimum of 50% informative reads derived from the paternal allele. This allelic bias was required to be reciprocal (in both directions of the crosses) and statistically supported with a P value < 0.01 (Pignatta et al. 2014). By applying these criteria, we could recover 25% (85 genes) and 44.6% (42 genes) of all comparable previously reported MEGs and PEGs, respectively (Fig. 4b; Dataset 1) (Wolff et al. 2011; Pignatta et al. 2014; Gehring et al. 2011; Hsieh et al. 2011). Previously published imprinting data were generated from hand-dissected endosperm at 6–8 DAP (Gehring et al. 2011; Hsieh et al. 2011; Pignatta et al. 2014), while our data were generated from 4 DAP endosperm. The developmental regulation of most imprinted genes is still unknown. Here, we identified more MEGs and PEGs than previously reported, suggesting that at earlier stages there are potentially more imprinted genes than at later stages of development. Consistently, PHE1 expression is strongly reduced at 5 DAP, which potentially impairs its detection as PEG after this stage. This, together with the high levels of tissue contamination in previously published samples, can explain the differences between our and published datasets.
In addition to canonical MEGs and PEGs (see above for definition), we defined potential MEGs and potential PEGs (poMEGs and poPEGs) as those genes that fulfilled all imprinting criteria in one direction of the crosses and were significantly parentally biased in the reciprocal cross, but the allelic bias was below the threshold of ≥ 85% maternal reads for MEGs and ≥ 50% paternal reads for PEGs (Table 1, Fig. 4a). Many previously reported MEGs and PEGs were found in this category (Fig. 4b), revealing that a fraction of potentially imprinted genes failed to be detected using our stringent criteria.
As a third category, we defined accession-specific imprinted genes as those genes that were only imprinted in one accession, but biallelically expressed in the other (Table 1). Epigenetic variation, such as presence/absence of transposable elements and associated accession-specific epigenetic modifications, explains accession-specific imprinting and several genes being imprinted in one accession but not in the other were previously reported (Pignatta et al. 2014). Only a small, but nevertheless significant subset of previously predicted accession-specific imprinted MEGs and PEGs overlap with our dataset (Fig. 4c). The low overlap is likely a consequence of different developmental stages and contamination levels between the samples.
As a fourth category, we defined genes with accession-specific biased expression in the endosperm. These genes show accession-biased expression independently of the direction of the cross (Table 1 and Fig. 4a). These genes may have an adaptive role in regulating seed development; or, alternatively, have close homologs with one of the homologs being active in one accession while inactive in the other. It is furthermore possible that those genes not being active in one of the accessions are on the evolutionary path to become pseudogenes. A significant subset of accession-specific imprinted genes identified in our dataset overlapped with those predicted in a previous study (Fig. 4d) (Pignatta et al. 2014); however, as indicated for the other categories, differences in stage and contamination levels likely account for the low level of overlap.
Together, our data show that INTACT-generated transcriptome profiles allow the reliable detection of parentally biased gene expression in the early endosperm. Our data furthermore suggest that imprinted genes are developmentally regulated, consistent with previous observations in maize (Xin et al. 2013). Consensus of criteria to define imprinted genes and reduced maternal tissue contamination by using the INTACT methodology are expected to overcome inconsistences in future datasets.
Conclusion
Cell type-specific transcriptome studies are of fundamental value to elucidate the development and function of specific cell types. Therefore, the application, development and constant improvement in methodologies enabling cell type-specific data collection are of immense value. Our study demonstrates that RNA-seq of nuclei isolated by INTACT is a reliable technique for endosperm-specific transcriptome studies and parent-of-origin-specific expression analyses. We anticipate that the development and adaptation of the INTACT method for nuclear transcriptome analyses of the endosperm will advance future studies focusing on the molecular events occurring in the endosperm. Furthermore, the use of endosperm-domain-specific promoters will allow dissecting the functional role of different domains of the endosperm. Despite its relevance for human nutrition and animal feed, we still lack a comprehensive understanding of the functional role of the endosperm for embryo development, the regulation and function of endosperm cellularization, and the functional role of the endosperm for seed coat development, just to name a few open knowledge gaps that remain to be addressed. We believe that the versatility of INTACT to isolate large numbers of pure endosperm nuclei that can be used for different types of downstream applications will help to address those questions and advance our understanding of the functional role of the endosperm.
References
Amin NM, Greco TM, Kuchenbrod LM et al (2014) Proteomic profiling of cardiac tissue by isolation of nuclei tagged in specific cell types (INTACT). Development 141:962–973. https://doi.org/10.1242/dev.098327
Barthelson RA, Lambert GM, Vanier C et al (2007) Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genom 8:340. https://doi.org/10.1186/1471-2164-8-340
Deal RB, Henikoff S (2010) A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell 18:1030–1040. https://doi.org/10.1016/j.devcel.2010.05.013
Deal RB, Henikoff S (2011) The INTACT method for cell type—specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat Protoc. https://doi.org/10.1038/nprot.2010.175
Feng J, Meyer CA, Wang Q et al (2012) GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28:2782–2788. https://doi.org/10.1093/bioinformatics/bts515
Gehring M, Missirian V, Henikoff S (2011) Genomic analysis of parent-of-origin allelic expression in Arabidopsis thaliana seeds. PLoS ONE 6:e23687. https://doi.org/10.1371/journal.pone.0023687
Goto K, Meyerowitz EM (1994) Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA. Genes Dev 8:1548–1560. https://doi.org/10.1101/gad.8.13.1548
Hatorangan MR, Laenen B, Steige KA et al (2016) Rapid evolution of genomic imprinting in two species of the Brassicaceae. Plant Cell 28:1815–1827. https://doi.org/10.1105/tpc.16.00304
Henry GL, Davis FP, Picard S, Eddy SR (2012) Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res 40:9691–9704. https://doi.org/10.1093/nar/gks671
Hsieh T-F, Shin J, Uzawa R et al (2011) Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci 108:1755–1762. https://doi.org/10.1073/pnas.1019273108
Jacob Y, Mongkolsiriwatana C, Veley KM et al (2007) The nuclear pore protein AtTPR is required for RNA homeostasis, flowering time, and auxin signaling. Plant Physiol 144:1383–1390. https://doi.org/10.1104/pp.107.100735
Köhler C, Page DR, Gagliardini V, Grossniklaus U (2005) The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nat Genet 37:28–30. https://doi.org/10.1038/ng1495
Krueger F, Andrews SR (2016) SNPsplit: allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Research 5:1479. https://doi.org/10.12688/f1000research.9037.2
Le BH, Cheng C, Bui AQ et al (2010) Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci USA 107:8063–8070. https://doi.org/10.1073/pnas.1003530107
Moreno-Romero J, Jiang H, Santos-González J, Köhler C (2016) Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm. EMBO J 35:1298–1311. https://doi.org/10.15252/embj.201593534
Moreno-Romero J, Santos-González J, Hennig L, Köhler C (2017) Applying the INTACT method to purify endosperm nuclei and to generate parental-specific epigenome profiles. Nat Protoc 12:238–254. https://doi.org/10.1038/nprot.2016.167
Moreno-Romero J, De Toro-De León G, Yadav VK et al (2018) Epigenetic signatures associated with imprinted paternally-expressed genes in the Arabidopsis endosperm. bioRxiv. https://doi.org/10.1101/423137
Palovaara J, Saiga S, Wendrich JR et al (2017) Transcriptome dynamics revealed by a gene expression atlas of the early Arabidopsis embryo. Nat Plants 3:894–904. https://doi.org/10.1038/s41477-017-0035-3
Pignatta D, Erdmann RM, Scheer E et al (2014) Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 3:e03198. https://doi.org/10.7554/eLife.03198
Przybyla D, Göbel C, Imboden A et al (2008) Enzymatic, but not non-enzymatic, 1 O 2 -mediated peroxidation of polyunsaturated fatty acids forms part of the EXECUTER1-dependent stress response program in the flu mutant of Arabidopsis thaliana. Plant J 54:236–248. https://doi.org/10.1111/j.1365-313X.2008.03409.x
Reynoso M, Pauluzzi G, Kajala K et al (2017) Nuclear transcriptomes at high resolution using retooled INTACT. Plant Physiol. https://doi.org/10.1104/pp.17.00688
Schon MA, Nodine M (2017) Widespread contamination of Arabidopsis embryo and endosperm transcriptome datasets. Plant Cell. https://doi.org/10.1105/tpc.16.00845
Steiner FA, Talbert PB, Kasinathan S et al (2012) Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res 22:766–777. https://doi.org/10.1101/gr.131748.111
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Villar C, Erilova A, Makarevich G, Trösch R, Köhler C (2009) Control of PHERES1 imprinting in Arabidopsis by direct tandem repeats. Mol Plant 2:654–660. https://doi.org/10.1093/mp/ssp014
Weinhofer I, Hehenberger E, Roszak P et al (2010) H3K27me3 profiling of the endosperm implies exclusion of Polycomb group protein targeting by DNA methylation. PLoS Genet 6:1–14. https://doi.org/10.1371/journal.pgen.1001152
Wolff P, Weinhofer I, Seguin J et al (2011) High-Resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLoS Genet 7:e1002126. https://doi.org/10.1371/journal.pgen.1002126
Xin M, Yang R, Li G et al (2013) Dynamic expression of imprinted genes associates with maternally controlled nutrient allocation during maize endosperm development. Plant Cell 25:3212–3227. https://doi.org/10.1105/tpc.113.115592
Yan D, Duermeyer L, Leoveanu C, Nambara E (2014) The functions of the endosperm during seed germination. Plant Cell Physiol 55:1521–1533. https://doi.org/10.1093/pcp/pcu089
Acknowledgements
This research was supported by a grant from the Knut and Alice Wallenberg Foundation (to C.K.) and an EMBO fellowship (to G.D.T-.D.L) (Grant No. EMBO ALTF 962-2017).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dolf Weijers.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A contribution to the special issue ‘Cellular Omics Methods in Plant Reproduction Research’.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Del Toro-De León, G., Köhler, C. Endosperm-specific transcriptome analysis by applying the INTACT system. Plant Reprod 32, 55–61 (2019). https://doi.org/10.1007/s00497-018-00356-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00497-018-00356-3