
Abstract
The syndrome of thrombotic microangiopathy (TMA) is a clinical-pathological entity characterized by microangiopathic hemolytic anemia, thrombocytopenia, and end organ involvement. It comprises a spectrum of underlying etiologies that may differ in children and adults. In children, apart from ruling out shigatoxin-associated hemolytic uremic syndrome (HUS) and other infection-associated TMA like Streptococcus pneumoniae-HUS, rare inherited causes including complement-associated HUS, cobalamin defects, and mutations in diacylglycerol kinase epsilon gene must be investigated. TMA should also be considered in the setting of solid organ or hematopoietic stem cell transplantation. In this review, acquired and inherited causes of TMA are described with a focus on particularities of the main causes of TMA in children. A pragmatic approach that may help the clinician tailor evaluation and management is provided. The described approach will allow for early initiation of treatment while waiting for the definitive diagnosis of the underlying TMA.
Similar content being viewed by others


Introduction
The assessment of an adult patient presenting with thrombotic microangiopathy (TMA) involves work-up for thrombotic thrombocytopenic purpura (TTP) due to deficiency of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin motif type 1, member 13), shigatoxin-induced hemolytic uremic syndrome (STEC-HUS), and secondary causes of TMA (infections, drugs, malignant hypertension, autoimmune diseases, and others). If other causes of TMA have been excluded, then complement-mediated/atypical hemolytic uremic syndrome (CM-HUS or aHUS) should be considered [1,2,3].
Although there is an overlap of the etiologies causing TMA in both adults and children, some of the diseases are more common in children and other rarer conditions are seen only in children. The main cause of TMA in children is STEC-HUS, followed by CM-TMA and Streptococcus-induced TMA (Sp-HUS) [4]. In addition, there are rare conditions such as congenital TTP, vitamin B12 metabolism defects (cobalamin C and G defects), and coagulation disorders (diacylglycerol epsilon mutation or variant DGKE) that may manifest as TMA, making the process of differential diagnosis more complex, especially in children younger than 2 years of age. Solid organ and hematopoietic stem cell transplantation (HSCT) may also present with TMA in children and may pose a challenge in differential diagnosis.
Although there are registries of specific diseases such as TTP and aHUS, TMA registries of young children are lacking. In a recent paper from Germany, among 232 patients with TMA, 61% presented with aHUS [5]. In a Brazilian aHUS cohort [6], among the 17 patients who presented with their first manifestation when less than 18 years old, 53% were younger than 2 years of age and one patient who was refractory to treatment with eculizumab had a homozygous mutation in DGKE [7].
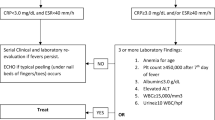
The possibility of various treatment strategies of these conditions underscores the need of a tailored diagnostic approach (Box). This review aims to discuss the causes of TMA—acquired and inherited (Fig. 1)—in children, and propose a practical algorithm to the diagnosis and management of TMA in this population (Fig. 2).

Acquired and inherited causes of TMA in children. The size of the balloons is proportional to the incidence of each disease. STEC-HUS shigatoxin-associated hemolytic uremic syndrome, Sp-HUS Streptococcus pneumoniae hemolytic uremic syndrome, DGKe diacylglycerol kinase epsilon, CM-HUS complement-mediated hemolytic uremic syndrome, iTTP immune-mediated thrombotic thrombocytopenic purpura, hTTP hereditary thrombotic thrombocytopenic purpura, TA-TMA transplant-associated TMA

Differential diagnosis of thrombotic microangiopathy in children: singularities of clinical presentation, tools for investigation and a pragmatic approach to management. MCV mean corpuscular volume, STEC-HUS shigatoxin-associated hemolytic uremic syndrome, Sp-HUS Streptococcus pneumoniae hemolytic uremic syndrome, CRP C-reactive protein, DGKe diacylglycerol kinase epsilon, CM-HUS complement-mediated hemolytic uremic syndrome, TTP thrombotic thrombocytopenic purpura, WES whole exome sequencing, ADAMTS13 a disintegrin and metalloprotease with thrombospondin type-1 repeats, 13thmember,TA-TMA transplant-associated TMA
Box—Checklist for diagnosis and evaluation of TMA in children
Identifying the presence of TMA |
• Complete blood count • Peripheral smear for evaluation of schistocytes • Reticulocyte count • Bilirubin • Lactic Dehydrogenase • Haptoglobin |
Evaluating the degree of organ injury |
• Complete physical exam including blood pressure and neurologic assessment • Kidney function: BUN, serum creatinine • Urinalysis and proteinuria • Liver enzymes • Pancreas enzymes: amylase, lipase • Blood bicarbonate and lactic acid levels • Troponin |
Defining underlying cause (request according to clinical suspicion) |
• Shigatoxin in stools • ADAMTS13 activity and inhibitor • Blood complement levels • Cultures: blood, urine, CRL, empyema • Latex test for Streptococcus pneumoniae • Tests for H1N1 and COVID19 • Other infections according to epidemiology: dengue, leptospirosis • Homocysteine and vitamin B12 blood levels • Urinary amino acid chromatography • Genetic testing: complement panel or Whole Exome Sequencing • Anti-factor H antibody • Kidney biopsy when secondary cause suspected or need to evaluate chronicity |
Shigatoxin-associated hemolytic uremic syndrome
Incidence and epidemiology
STEC-HUS is one of the most common causes of acute kidney injury in children [8, 9]. The disease was first reported in 1955 by Gasser et al. [10]. Diarrhea associated with shigatoxin-producing Escherichia coli is estimated to occur in 2.8 million cases/year worldwide and the incidence of STEC-HUS varies from 0.5 cases/100,000 population in Finland [11] to 12 cases/100,000 population in Argentina, where it is endemic [12]. Seasonal variations occur, with a greater number of cases in the summer. It is more common in children under 5 years of age and E. coli O157 is the most common strain producing shigatoxin in this age group in the USA [13]. In a cohort from Finland [11], age under 3 years was a risk factor for HUS in STEC-positive children. In 2011, the largest outbreak to date occurred in Germany with almost 4000 cases of diarrhea due to E. coli O104, with 845 cases of STEC-HUS and 54 deaths. There were 90 cases in children with one death and the outcomes were similar to previous case series, although mean age was higher (most children were older than 10 years) [14]. In Europe, non-O157 serotypes of E. coli have become more common [15]. In a prospective study in northern Italy, incidence of STEC-HUS among patients with shigatoxin-positive bloody diarrhea stool tests was 15.9% [16].
Clinical presentation and diagnosis
STEC-HUS is characterized by platelet consumption followed by hemolysis and acute kidney injury. Clinically, a prodrome of profuse, bloody diarrhea occurs about 2 to 5 days after STEC infection. The signs and symptoms of microangiopathy occur a few days after diarrhea, but only in 10 to 20% of patients. Extrarenal manifestations may involve central nervous, cardiovascular, gastrointestinal (including pancreas), and musculoskeletal systems [17]. In addition to the platelet consumption, hemolysis, and kidney injury that characterize TMA, there may be a decrease in the blood level of component 3 (C3) of the complement. In rare cases, a decrease in C4 occurs. This finding is usually present in the most severe cases [18, 19] and is temporary—and may be due to a secondary involvement of the complement system in vascular inflammation [20]. Therefore, the presence of low C3 in patients with TMA does not exclude the possibility of STEC-HUS [21]. Increase in inflammatory markers such as C-reactive protein and d-dimer is frequent in patients with STEC-HUS [22]. It is essential that the collection of fecal material for stool culture or PCR for shigatoxin be done early on admission since positivity decreases after the 10th day of diarrhea, and after 15 days 90% of patients present with negative stool cultures in a prospective cohort from Argentina [23]. We reiterate that it is essential to collect material for shigatoxin in all cases of TMA and to discuss with local health authorities the availability of sending material for the detection of shigatoxin in feces in specialized laboratories. When available, IgA, IgG, and IgM antibodies to E. coli lipopolysaccharide (LPS) can be detected later in the course of the disease especially when stool specimens fail to detect the presence of shigatoxin [15]. Specifically for E. coli O157 serotype, the Glyco-iELISA to detect IgM antibodies has significantly increased accurate diagnosis of STEC-HUS [24].
When recovery of STEC-HUS extends beyond 2 weeks, the differential diagnosis with atypical HUS should be considered. Clinical and laboratory findings that make STEC-HUS less likely [84]:
-
– Negative stool culture or PCR-shigatoxin in feces (harvested up to 10 days after diarrhea)
-
– Persistent thrombocytopenia beyond the first week in the absence of a superimposed infectious condition
-
– Relapsing pattern of TMA after resolution of initial manifestation
-
– Kidney injury that persists for more than 4 to 6 weeks in the absence of superimposed septic or hypovolemic shock
-
– Absence of diarrhea and/or negative shigatoxin test
-
– Persistently low C3
In these situations, the main differential diagnosis, especially in the pediatric age group, is aHUS [4], which reinforces the importance of early collection of specimens for shigatoxin or stool culture (where shigatoxin is not available) at the admission of ALL patients with TMA.
Genetic variants in alternative complement genes have been reported in patients with STEC-HUS [25] and should be suspected in patients who progress to kidney failure or have recurrent TMA or persistently low C3.
Pathophysiology
Karmali et al. [26] were the first to describe the association between gastrointestinal infections by shigatoxin-producing E. coli (Stx; verotoxin) and STEC-HUS. Since then, these strains have been described as a major cause of STEC-HUS in both sporadic cases and in outbreaks in many countries, usually associated with contamination of water, vegetables, beef products, and other foods [9]. HUS–E. coli strains produce shigatoxin 1 (Stx1) and/or shigatoxin 2 (stx2). Shigatoxins are 70 kd holotoxins that contain a single A subunit and 5 B subunits. Cell damage results when the B subunit recognizes and binds to globotriaosylceramide (Gb3 or CD77), which resides in the plasma membrane of certain eukaryotic cells including the kidney and brain endothelial cells. Cellular lesions were recently described by Volokhina et al. [27], although the exact mechanism of injury is still unknown. The toxin–Gb3 complex is internalized and enters the cytoplasm, where subunit A is synthesized. Cellular apoptosis results from Gb3 binding with endocytosis, retrograde transport, cytosolic translocation of shigatoxin, and consequent ribosomal inactivation, which leads to endothelial damage, exposure of the subendothelial layer and consequent TMA. The toxin also acts on cell activation, pro-inflammatory and pro-thrombotic pathways, facilitating thrombosis by von Willebrand factor endothelial secretion [8].
Management
There is no specific treatment for STEC-HUS. Clinical care involves supportive measures with a focus on stabilizing the patient to natural resolution and recovery. Early volume expansion has been demonstrated to improve neurological and kidney outcomes of patients with STEC-HUS (weight gain of 12.5% vs. 0%) [28]. A systematic review has shown that no therapy tested to date has shown a greater benefit than supportive treatment [13]. An analysis of the German outbreak—the biggest to date that happened in 2011 with more than 3000 cases of diarrhea and 800 cases of STEC-HUS [29, 30]—showed that the average duration of diarrhea was 9.3 days (median 6 days) and most patients were admitted to the hospital in the first week after the onset of diarrhea. In the German outbreak, plasmapheresis [31] and corticosteroids showed no benefit, as well as the use of the terminal complement blocker eculizumab (Soliris, Alexion Pharma) [32], although results from clinical trials with eculizumab in STEC-HUS are still awaited. For a comprehensive review of eculizumab in STEC-HUS, we recommend the paper from Walsh and Johnson [33]. More recently, a case report described good outcomes with pulse steroids in a 7-year-old patient with STEC-HUS and late neurologic involvement [22, 34]. Fosfomycin used in the first 2 or 3 days after onset of diarrhea may be beneficial in preventing the evolution to STEC-HUS [35]. In the evolution of STEC-HUS, platelet count was the first to improve (end of the first week), followed by improvement of the hemoglobin and slow drop in lactate dehydrogenase. The acute kidney injury took an average of 2 weeks to recover, starting with a return of diuresis and then a fall in blood urea nitrogen and creatinine. Around 50% of patients required kidney replacement therapy and only three out of 160 patients were dependent on chronic dialysis. However, the kidney injury may be longer in very severe cases (LMPP and MHVG have seen cases that took up to 5 weeks before improvement in kidney function). Approximately 30% of patients with STEC-HUS continue to have variable degrees of neurological or kidney sequelae [13]—proteinuria and systemic arterial hypertension. Therefore, it is recommended that every patient with STEC-HUS be followed by a nephrologist or pediatric nephrologist after hospital discharge. There may be a role for probiotics in the management and prevention of STEC-HUS [36], but further studies are warranted.
Complement-mediated, atypical hemolytic uremic syndrome
Incidence and epidemiology
Atypical hemolytic uremic syndrome (aHUS) is a cause of TMA driven by an alternative complement pathway dysregulation. More recently, the term complement-mediated hemolytic uremic syndrome (CM-HUS) has been advocated since complement overactivation has been found in conditions otherwise considered secondary TMA (as in malignant hypertension, for instance), and this may have important therapeutic implications [37]. For the purposes of this review, aHUS is used for cases of CM-TMA without any other underlying secondary condition (i.e., primary CM-TMA). It is classified as an ultrarare disease with an incidence of less than 1 case per million population per year [38]. Up to 50–60% of patients present with a genetic defect or autoantibody leading to overactivation of alternative complement pathway, but positivity of genetic findings is variable worldwide—from 18% in a cohort from India [39] to 80% in Japan [40]. Progression to kidney failure or death had a significant decrease after introduction of terminal complement inhibitor eculizumab: from 30–50 to 9% in children, and from 56–67 to 6–15% in adults [41].
Clinical presentation and diagnosis
aHUS is characterized clinically by the TMA triad: non-immune microangiopathic hemolytic anemia, thrombocytopenia or a significant (> 25%) drop in platelet count, and evidence of organ injury in variable degrees. Since there is no gold standard diagnostic test for aHUS, it is important to rule out other more common causes of TMA [42], which is a challenge in a disease with high morbidity and mortality [43]. One of the key questions in aHUS is the correlation of genetic findings with clinical presentation. In the great majority of patients, genetic variants present in heterozygosis with incomplete penetrance, frequently associated with one or more risk polymorphisms, and an environmental factor may be necessary to result in full blown TMA clinical presentation. In 2007, the French Society of Pediatric Nephrology published data of 46 children with aHUS and found that 52% had a mutation, with CFH mutation carrying a worse prognosis and poor transplant outcomes [44]. In 2010, data analysis from 273 patients enrolled in the International Registry of Recurrent and Familiar HUS/TTP concluded that adults had a worse prognosis than children, as well as familial cases when compared to sporadic cases. Overall, 75% of patients presented either a mutation or a risk polymorphism, but there was an ethnicity-specific genotype [43]. Bresin et al. [45] found that risk haplotypes in CFH and MCP, when combined with mutations, carry a worse outcome. More recently, data from The Global aHUS Registry with more than 800 patients enrolled confirmed that adults have worse kidney outcome than children, as well as patients carrying CFH pathogenic variants [46].
Pathophysiology
aHUS is a rare disease caused in most cases by a dysregulation of the alternative complement pathway (Fig. 3). The first association between aHUS and the alternative complement defect was described in 1981 [47], when siblings with aHUS and consanguineous parents were found to have decreased blood levels of CFH (Complement Factor H). In 1998, the team lead by T. Goodship [48] unraveled the association between aHUS and chromosome 1q32, which contains the genes for complement regulators. In the following years, defects in other components of complement regulators of the alternative pathway have been successively found—lack of function mutations in CD46 (MCP—membrane cofactor protein), CFI (complement factor I), thrombomodulin (THBD), CFHR1–5 (proteins related to factor H 1 to 5 and hybrid genes), CFP (complement factor properdin), and gain of function mutations in genes encoding C3 convertase components, CFB (complement factor B) and C3. The role of anti-factor H antibodies in the pathogenesis of aHUS, especially in children, was also demonstrated and its detection has therapeutic implications [49]. It is very important to determine the underlying genetic status since it adds to prognosis and management. It is well known that patients with factor H pathogenic variants have worse outcomes and higher rates of post kidney transplant recurrence, and patients with MCP variants or high-risk polymorphisms rarely have aHUS recurrence after transplant, although they might experience recurrent bouts of TMA throughout their life [46, 50, 51].

Complement-mediated TMA in a child less than 2 years of age. A Light microscopy showing glomeruli with mesangiolysis, double contouring of the capillary walls, and lobular accentuation of the glomerular capillary tufts (Periodic acid Schiff stain 60 ×). B Light microscopy showing arterioles with endothelial swelling and near complete occlusion of arteriolar lumen (Periodic acid Schiff stain, 60 ×). C, D Electron microscopy showing capillary wall injury with subendothelial expansion by fluffy granular material, injured cellular elements, and new basement formation resulting in double contours (C: 2500 × , D: 8000 ×). Asterix indicates mesangiolysis, white arrows indicate arteriolar occlusion by TMA, and black arrows indicate double contour formation
Management
Besides understanding the pathophysiology of aHUS, determining the underlying defect of the alternative pathway is key to address specific treatment, although it is not necessary to have the result of genetic tests to start treatment. In September 2011, eculizumab (Soliris, Alexion) was approved by the Food and Drug Administration (FDA, USA) as the first terminal complement inhibitor for use in patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) and aHUS. The outcomes of patients with aHUS treated with eculizumab have been published in pivotal trials both in adults [52,53,54] and children [55], and were extensively reviewed [56]. Almost a decade after the first published use of eculizumab for aHUS [57], the question is whether all patients need lifelong treatment and, if not, who would be eligible to stop. Results of a prospective study on discontinuation of eculizumab [58] in 57 patients from 22 centers in France were published. In this trial, patients were on eculizumab for at least 6 months and in remission at the time of discontinuation. Importantly, patients should have (1) glomerular filtration rate (GFR) > 60 mL/min/1.73 m2 (Schwartz or MDRD formulas for children and adults, respectively) and urinary protein/creatinine ratio (UPC) < 0.05; or (2) stable kidney function in the past six months. The relapse rate was higher in children than adults (31% vs. 19%, respectively) and in patients who carried a pathogenic genetic finding, except for one patient with a negative genetic test—a more detailed genetic analysis revealed a homozygous mutation in the ADAMTS13 gene and plasma activity of ADAMTS13 was decreased. At the end of the second year of follow-up, patients who had aHUS recurrence evolved with greater proteinuria than those who did not relapse. Although the authors concluded that stopping eculizumab in controlled patients is safe, the risk of relapse needs to be discussed with patients and caregivers and eculizumab should be promptly available if necessary. In addition to eculizumab, a newer formulation of long-acting C5 inhibitor ravulizumab is now available for treatment of aHUS; data from phase 3 trials have demonstrated good efficacy and safety results in adult and pediatric patients [59, 60].
Streptococcus pneumoniae-associated hemolytic uremic syndrome
Incidence and epidemiology
Streptococcus pneumoniae-associated hemolytic uremic syndrome (Sp-HUS) is an important cause of TMA in young children. It accounts for 5–15% of cases of HUS in children (1–3% of all causes of TMA) with increasing prevalence probably due to serotypes not covered by the vaccine [61]. The highest incidence occurs in children under 2 years of age [62] who have Sp-pneumonia/empyema (60%) or meningitis (30%). The incidence of pneumococcal infections that progress with HUS ranges from 0.4 to 0.6% [63]. Prior to 1990, there was a 50% mortality and 67% of the patients who survived progressed to chronic kidney disease (CKD) or hypertension. In 2007, mortality and progression to CKD or hypertension decreased to 12.3% and 16%, respectively, likely due to increased awareness and management in the intensive care unit.
Clinical presentation and diagnosis
The clinical presentation is that of classic TMA triad, occurring usually 7 to 9 days after the onset of pneumococcal infection. The differential diagnosis includes disseminated intravascular coagulation (DIC) secondary to pneumococcal infection, which presents with features of TMA but also bleeding and altered prothrombin time and activated partial thromboplastin time. Evolution of anuria/oliguria and thrombocytopenia is more prolonged than with shigatoxin-associated HUS (STEC-HUS) [61]. The direct Coombs test, unlike all other causes of TMA, is positive in 90% of cases and aids in the diagnosis of Sp-HUS [61].
Pathophysiology
Pneumococci release an enzyme called neuraminidase which removes neuraminic acid and exposes the Thomsen–Friedenreich antigen (T-antigen) present on red blood cells, platelets, and glomerular endothelial cells [64, 65]. The T-antigen is recognized by preformed IgM cold antibodies. More recently, decrease in sialylation of transferrin and IgA1 O-glycans was shown and may add to the pathophysiology of Sp-HUS, which is still not completely understood. In the early 2000s, studies showed that pneumococcal neuroaminidase leads to a disruption in Factor H, making it difficult to bind to C3 convertase effectively, culminating in the activation of the alternative complement pathway and cell injury [66].
Management
Treatment of Sp-HUS is based on supportive therapy with dialysis and initiation of antibiotic therapy. Early detection and rapid onset of antibiotics decrease mortality. In the most severe cases, use of the combination of vancomycin and cephalosporins to broaden the bacterial spectrum should be considered. Based on the cultures and sensitivity tests available, vancomycin should be discontinued, and therapy directed according to the results. The duration of treatment depends on the site of pneumococcal infection; if there is a need for platelet transfusions or red blood cell concentrate, they should be washed to remove IgM antibodies against Thomsen–Friedenreich antigen. The removal of neuroaminidase by plasma exchange is hypothetically indicated. However, plasma exchange is controversial since IgM antibody against the Thomsen–Friedenreich antigen may result in possible increased polyagglutination and aggravation of the microangiopathic phenomenon [62]. There are case reports of the use of complement inhibitor eculizumab in Sp-HUS with good response [67].
Immune-mediated (acquired) thrombotic thrombocytopenic purpura
Incidence and epidemiology
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is an immunological disorder caused by IgG autoantibodies that inhibit the function of ADAMTS13, which is to cleave the ultralarge multimers of von Willebrand Factor on endothelial cells. Although its incidence is higher in adults than in children, it is a severe condition that needs prompt recognition and treatment.
Clinical presentation and diagnosis
The classic clinical pentad of iTTP comprises hemolytic anemia, thrombocytopenia, neurological symptoms, jaundice and fever, although only a minority present with all symptoms [68]. In order to differentiate TTP from other causes of TMA in the intensive care unit, the Harvard TMA Research Collaborative developed a diagnostic score for TTP called Plasmic Score [69] which includes platelet count, hemolysis, no active cancer, no history of transplant, mean corpuscular volume of red cells < 90 fL, normal coagulation, and creatinine < 2.0 mg/dL (176 mmol/L). The score can be used on the bedside and has a high predictive value for severe ADAMTS13 deficiency.
Pathophysiology
The basic etiology was discovered in the early 1980s, when giant von Willebrand factor multimers (ULVWF, Ultra Large von Willebrand Factor) were found in the plasma of patients with TTP [70]. ULVWF maintain primary hemostasis, promote platelet adhesion, and aggregation through binding to platelet glycoproteins (GP) 1b–IX and GP IIbIIIa [71]. In addition, ULVWF binds to free FVIII in the plasma, protecting it against proteolysis. ULVWF is stored mainly in the Weibel Palade bodies of endothelial cells and secreted into plasma in the form of large multimers. ADAMTS13 degrades these ULVWF high-density multimers, cleaving the A2 domain 842Tyr–843Met peptide bond, generating multimers ranging in size between 500 and 20,000 kDa [72]. The action of ADAMTS13 is increased in places or situations where there is greater stress on blood flow, that is, where the ratio between the pressure of blood flow and the diameter of the vessel is high [73]. Severe ADAMTS13 deficiency (defined as less than 10% of activity) results in unusual, giant FVW multimers at risk of platelet thrombus in small vessels causing red blood cell “shear” and occlusion of vessels with tissue ischemia. The HLA variant rs6903608 has been shown to be a risk factor for both immune TTP onset and relapse in an Italian cohort and may serve as a biomarker for worse outcome [74].
Management
Without treatment, iTTP has a 95% mortality rate with most deaths occurring within 24 h after initial presentation [75]. The recognition of iTTP in the first hours (“at bedside”) is crucial to institute treatment. If the collection of ADAMTS13 activity is feasible, it must be done (in a citrated tube) before beginning plasma exchange. It is worth mentioning that collecting or waiting for the result of the ADAMTS13 test should not delay the start of treatment, which is a priority. The use of plasma exchange leads to a response rate of 80% and survival greater than 90% when initiated in the first 4 to 8 h of suspected diagnosis and the American Society for Apheresis (ASFA) [76] has classified the procedure as indication level I for iTTP. When unavailable, plasma can be infused in a volume of 25 to 30 mL/kg of fresh plasma until plasmapheresis is available, although a randomized trial showed plasma infusions to be less effective in reducing mortality [77]. In addition, prednisone 1 mg/kg/day or pulse therapy with methylprednisolone should be combined. The current recommendation is daily plasma exchange until platelets are above 150,000/mm3, when plasma exchange can be stopped [78]. Solvent/detergent-treated (inactivates lipid-enveloped viruses) pooled plasma (OCTAPLAS) may be used in children instead of fresh frozen plasma if available [79]. Recently, a new medication for the treatment of iTTP has been developed. Caplacizumab is a nanobody that binds to von Willebrand factor preventing platelet adhesion. In two prospective studies that enrolled 220 adults, patients who received caplacizumab together with plasma exchange and immunosuppression had lower rates of acute disease exacerbation [80, 81] and no mortality, which led to the drug being FDA and EU-approved. Caplacizumab use together with steroids and rituximab was retrospectively analyzed in 4 children (among 85 patients) in the UK—overall, the bleeding rate was 55%, thrombotic events occurred in 16%, and five patients died (four of whom with the drug starting 48 h after plasma exchange). Since thrombotic events are much more frequent than hemorrhage, platelet transfusions should be avoided except in cases of severe bleeding [76].
Hereditary thrombotic thrombocytopenic purpura
Incidence and epidemiology
Hereditary thrombotic thrombocytopenic purpura (hTTP), also known as Upshaw–Shulman Syndrome [82], is rare (10% of cases of TTP) and caused by homozygous mutations or by compound heterozygous mutations of the ADAMTS13 gene; therefore, it is more prevalent in offspring of consanguineous parents. Patients with simple heterozygous mutations have no apparent abnormalities. There is a bimodal distribution according to age of diagnosis, with 50% presenting at age 2–5 years and 50% in young women usually triggered by pregnancy [78].
Clinical presentation and diagnosis
The clinical features of the hereditary form are recurrent episodes of microangiopathic hemolytic anemia and thrombocytopenia, usually with neurological manifestations or other signs of organ damage. Its diagnosis requires confirmation of severe ADAMTS13 deficiency (plasma activity < 10%) in the absence of inhibitory antibodies (non-inhibitory antibodies may be present) and confirmed by finding a pathogenic mutation in the ADAMTS13 gene. There is often a delay in diagnosis of hTTP; the Hereditary TTP Registry [83] has enrolled more than 120 patients with mean age of presentation 4.5 years and mean age of diagnosis 16.7 years, showing the gap in knowledge regarding this disease.
Pathophysiology
Compared to iTTP, rather than decreased ADAMTS13 activity due to antibodies targeting ADAMTS13, in hTTP, ADAMTS13 (antigen) is deficient due to an underlying genetic abnormality in the ADAMTS13 gene. When ADAMTS13 is lacking, ultra-large vWF multimers persist in the circulation resulting in aggregation of platelets, which are activated and form the vWF-rich microthrombi (platelet rich microthrombi are a hallmark of TTP). This process causes microthrombi and occlusion of microvessels leading to organ ischemia. Resultant laboratory features include thrombocytopenia, and evidence of mechanical destruction of red blood cells (schistocytes and hemolytic anemia). Levels of ADAMTS13 are variable and dependent on the underlying genetic variant; this has an impact on severity and presentation [78].
Management
Current treatment is plasma infusion 10 to 15 mL/kg every 21–28 days.
TMA mediated by cobalamin defects
Incidence and epidemiology
Patients with an inherited vitamin B12 disorder are at risk for developing TMA. It has been mainly observed in neonates or in infants during the first year of life with methylmalonic aciduria and homocystinuria, caused by cbl deficiency type C (cblC; OMIM #277,400) [84]. Cbl C disease mostly manifests as failure to thrive, hypotonia, seizures, microcephaly, and delayed development. Three cases were diagnosed in Brazil, one of them was published in which the whole exome sequencing revealed two novel likely pathogenic heterozygous variants in MTR which encodes for methionine synthase, suggesting the diagnosis of methionine synthase deficiency (MSD) cbl C disease-mediated TMA [85]. The suspicion was made due to the lack of clinical and laboratory response to the use of eculizumab. This patient had mild neurological impairment that was characterized by developmental delay, mainly his speech. Few adult cases have previously been reported [86, 87].
Clinical presentation and diagnosis
Clinically, patients with a defect in cobalamin metabolism manifest with failure to thrive, hypotonia, seizures, microcephaly, and several degrees of delayed development. Neurological sequelae are common in affected children. Adult-onset disease is rare and may present with neurological manifestations such as cognitive impairment, ataxia, and psychosis. Cbl deficiency, or a defect in its metabolism, result in hyperhomocystinemia, decreased plasma methionine level, and methylmalonic aciduria. The reported adults had microangiopathic anemia, thrombocytopenia, acute kidney injury, and hypertension. Chronic kidney disease with hypertension and proteinuria have been reported in up to 40% of patients.
Pathophysiology
Homozygous or heterozygous mutations in MMACHC (chromosome 1p34.1) cause deficiency of the Cbl-C enzyme. The Cbl-C enzyme is responsible for the metabolization of cobalamin into methylcobalamin and adenosylcobalamin. Furthermore, the lack of Cbl-C enzyme leads to the accumulation of homocysteine and methylmalonic acid in endothelial cells, which promote increased platelet aggregation, increased free radicals, increased expression of local procoagulant factors, and induce the binding of tissue plasminogen activator to the endothelium. All of these factors result in endothelial damage and trigger intravascular thrombosis with microangiopathic anemia, thrombocytopenia, and ischemia in organs. The resulting deficiency of methylcobalamin leads to hyperhomocysteinemia in the presence of normal blood levels of vitamin B12 [1].
Management
The main treatment for infants is parenteral hydroxycobalamin [86] (1 mg IM daily), which prevents progression to kidney failure and microangiopathic manifestations. Therefore, we suggest that homocysteine and vitamin B12 in the blood should be performed in all patients with TMA, especially if there is a neurological condition, persistently high levels of lactic dehydrogenase, average corpuscular volume > 100 microns3, or resistance to treatment with complement inhibitor.
TMA-mediated by diacylglycerol kinase epsilon mutation
Incidence and epidemiology
The Mendelian forms of HUS have been associated with complement genes, mostly transmitted as autosomal dominant variants (variable penetrance) and more rarely, autosomal recessive mutations (high penetrance). In 2013, recessive mutations in diacylglycerol kinase epsilon mutation (DGKE), a non-complement gene, were described in families with TMA through whole exome sequencing [88]. The incidence is still unknown, but it is an ultrarare cause of TMA.
Clinical presentation and diagnosis
Patients with recessive loss-of-function mutations in DGKE gene have phenotypes ranging from membranoproliferative glomerulonephritis, steroid-resistant-nephrotic syndrome [89] to HUS [90]. Usually, patients have hypertension and hematuria and the advance to CKD is slow and progressive. The search for a mutation in the DGKE gene is indicated in cases of TMA beginning in the first year of life—especially if there is significant proteinuria and the parents are consanguineous—and in cases where the response to terminal complement inhibition is unsatisfactory.
Pathophysiology
Diacylglycerol kinase (DGK) is a family of enzymes that catalyze the conversion of diacylglycerol (DAG) to phosphatidic acid (genecards.org)—both thought to function as bioactive lipid signaling molecules with important roles in the biosynthesis of complex lipids. Currently, nine members of the DGK family have been identified. DGKE has specificity for arachidonate-containing DAG. A type of TMA due to recessive mutations in the DGKE gene has been described [88]—loss of activity of the enzyme DAG, present in endothelial cells, platelets, and podocytes, induces apoptosis in endothelial cells and impairs angiogenic response, leading to a prothrombotic and inflammatory state with consequent TMA. Although the role of complement in kidney disease in patients with DGKE mutations was initially neglected, some patients with concomitant mutations in thrombomodulin and C3 have been identified and this possibly explains the spectrum of clinical manifestations in these patients, which normally occur as early as the first year of life. This type of isolated DGKE mutation is not associated with post-kidney transplant recurrence.
Management
The benefits of plasma exchange or plasma infusion and immunosuppression have been inconsistent. Recurrences occurred in patients who were receiving eculizumab [42] and recently a Brazilian case was published in which the detection of DGKE mutation was made in an adult due to non-response to eculizumab [7]. The treatment so far is restricted to nephroprotective measures (strict control of blood pressure, blockade of the renin–angiotensin–aldosterone system, metabolic control).
Transplant-associated TMA
Transplant-associated TMA (TA-TMA) results from endothelial damage occurring in recipients of solid organ or hematopoietic stem cell transplantation (HSCT). The pathophysiology of this entity is complex and multifactorial. In solid organ transplant recipients, the main differential diagnoses are ischemia–reperfusion injury, calcineurin-inhibitor toxicity, antibody-mediated rejection, infections, recurrence of aHUS, and STEC-HUS. It is important to rule out these entities and consider complement inhibition early if aHUS recurrence is suspected [91]. Among patients with allogenic HSCT, TMA may result from multiple mechanisms of endothelial injury that include infections, calcineurin inhibitors, graft versus host disease, and drugs. The cumulative incidence among allogenic HSCT is 3% [92]. There may be underlying genetic variants in complement genes and the experience of a large cohort published by Jodele et al. [93] showed a significant improvement in 1-year post-HSCT survival from 16.7 to 66% with short course eculizumab use (median 11 infusions). Interestingly, TA-TMA usually presents with hemolysis and platelet consumption that is preceded by hypertension, with only mild and late kidney involvement.
Conclusion
Diagnosis and uncovering the etiology of TMA in children is of utmost importance in management of the disease entity. The causes of TMA in children less than 2 years old is variable and includes the more common entities of STEC-HUS and aHUS; the less common entities such as sp-HUS, iTTP, and hTTP; and rare entities including cblC-TMA and DGKE-TMA. TMA in the setting of solid organ or HSC transplantation has a multifactorial pathophysiology and children may have additional underlying complement defects. It is critical to recognize and correctly diagnose the specific TMA entities since the management, outcomes, and prognosis depend on early diagnosis and institution of appropriate treatment.
References
George JN, Nester CM (2014) Syndromes of thrombotic microangiopathy. N Engl J Med 371:654–666
Laurence J, Haller H, Mannucci PM, Nangaku M, Praga M, Rodriguez de Cordoba S (2016) Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol 14(Suppl 11):2–15
Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, Lammle B, Matsumoto M, Pavenski K, Sadler E, Sarode R, Wu H, International Working Group for Thrombotic Thrombocytopenic Purpura (2017) Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost 15:312–322
Joly BS, Zheng XL, Veyradier A (2018) Understanding thrombotic microangiopathies in children. Intensive Care Med 44:1536–1538
Schonermarck U, Ries W, Schroppel B, Pape L, Dunaj-Kazmierowska M, Burst V, Mitzner S, Basara N, Starck M, Schmidbauer D, Mellmann A, Dittmer R, Jeglitsch M, Haas CS (2020) Relative incidence of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome in clinically suspected cases of thrombotic microangiopathy. Clin Kidney J 13:208–216
Palma LMP, Eick RG, Dantas GC, Tino M, de Holanda MI, Brazilian Thrombotic M, Atypical Hemolytic Uremic Syndrome Study Group (2021) Atypical hemolytic uremic syndrome in Brazil: clinical presentation, genetic findings and outcomes of a case series in adults and children treated with eculizumab. Clin Kidney J 14:1126–1135
de Holanda MI, Gomes CP, Araujo SA, Wanderley DC, Eick RG, Dantas GC, Tino M, Pesquero JB, Palma LMP (2019) Diacylglycerol kinase epsilon nephropathy: late diagnosis and therapeutic implications. Clin Kidney J 12:641–644
Andreoli SP, Trachtman H, Acheson DW, Siegler RL, Obrig TG (2002) Hemolytic uremic syndrome: epidemiology, pathophysiology, and therapy. Pediatr Nephrol 17:293–298
Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB (2002) Clinical course and the role of shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis 186:493–500
Gasser C, Gautier E, Steck A, Siebenmann RE, Oechslin R (1955) Hemolytic-uremic syndrome: bilateral necrosis of the renal cortex in acute acquired hemolytic anemia. Schweiz Med Wochenschr 85:905–909
Ylinen E, Salmenlinna S, Halkilahti J, Jahnukainen T, Korhonen L, Virkkala T, Rimhanen-Finne R, Nuutinen M, Kataja J, Arikoski P, Linkosalo L, Bai X, Matussek A, Jalanko H, Saxen H (2020) Hemolytic uremic syndrome caused by Shiga toxin-producing Escherichia coli in children: incidence, risk factors, and clinical outcome. Pediatr Nephrol 35:1749–1759
Luna M, Kamariski M, Principi I, Bocanegra V, Valles PG (2020) Severely ill pediatric patients with Shiga toxin-associated hemolytic uremic syndrome (STEC-HUS) who suffered from multiple organ involvement in the early stage. Pediatr Nephrol 36:1499–1509
Keir LS (2015) Shiga toxin associated hemolytic uremic syndrome. Hematol Oncol Clin North Am 29:525–539
Kemper MJ (2012) Outbreak of hemolytic uremic syndrome caused by E. coli O104:H4 in Germany: a pediatric perspective. Pediatr Nephrol 27:161–164
Rastawicki W, Smietanska K, Rokosz-Chudziak N, Wolkowicz T (2020) Antibody response to lipopolysaccharides and recombinant proteins of Shiga toxin (STX)-producing Escherichia coli (STEC) in children with haemolytic uraemic syndrome in Poland. Lett Appl Microbiol 70:440–446
Ardissino G, Possenti I, Vignati C, Daprai L, Capone V, Brigotti M, Luini MV, Consonni D, Montini G (2020) Is Shigatoxin 1 protective for the development of Shigatoxin 2-related hemolytic uremic syndrome in children? Data from the ItalKid-HUS Network. Pediatr Nephrol 35:1997–2001
Khalid M, Andreoli S (2019) Extrarenal manifestations of the hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli (STEC HUS). Pediatr Nephrol 34:2495–2507
Netti GS, Santangelo L, Paulucci L, Piscopo G, Torres DD, Carbone V, Giordano P, Spadaccino F, Castellano G, Stallone G, Gesualdo L, Chironna M, Ranieri E, Giordano M (2020) Low C3 serum levels predict severe forms of STEC-HUS with neurologic involvement. Front Med (Lausanne) 7:357
Balestracci A, MeniBataglia L, Toledo I, Beaudoin L, Alvarado C (2020) C3 levels and acute outcomes in Shiga toxin-related hemolytic uremic syndrome. Pediatr Nephrol 35:331–339
Westra D, Volokhina EB, van der Molen RG, van der Velden TJ, Jeronimus-Klaasen A, Goertz J, Gracchi V, Dorresteijn EM, Bouts AH, Keijzer-Veen MG, van Wijk JA, Bakker JA, Roos A, van den Heuvel LP, van de Kar NC (2017) Serological and genetic complement alterations in infection-induced and complement-mediated hemolytic uremic syndrome. Pediatr Nephrol 32:297–309
Noris M, Mescia F, Remuzzi G (2012) STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 8:622–633
Joseph A, Cointe A, MarianiKurkdjian P, Rafat C, Hertig A (2020) Shiga Toxin-associated hemolytic uremic syndrome: a narrative review. Toxins (Basel) 12:67
Lucarelli LI, Alconcher LF, Arias V, Galavotti J (2021) Duration of fecal shedding of Shiga toxin-producing Escherichia coli among children with hemolytic uremic syndrome. Arch Argent Pediatr 119:39–43
Wijnsma KL, Veissi ST, van Bommel SAM, Heuver R, Volokhina EB, Comerci DJ, Ugalde JE, van de Kar N, van den Heuvel L (2019) Glyco-iELISA: a highly sensitive and unambiguous serological method to diagnose STEC-HUS caused by serotype O157. Pediatr Nephrol 34:631–639
Celakil ME, Yucel BB, Bek K (2020) CFH and CFB mutations in Shiga toxin-associated haemolytic uraemic syndrome in a 6-year-old boy. Paediatr Int Child Health 40:129–131
Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H (1985) The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis 151:775–782
Volokhina EB, Feitz WJC, Elders LM, van der Velden T, van de Kar N, van den Heuvel L (2020) Shiga toxin selectively upregulates expression of syndecan-4 and adhesion molecule ICAM-1 in human glomerular microvascular endothelium. Toxins (Basel) 12:435
Ardissino G, Tel F, Possenti I, Testa S, Consonni D, Paglialonga F, Salardi S, Borsa-Ghiringhelli N, Salice P, Tedeschi S, Castorina P, Colombo RM, Arghittu M, Daprai L, Monzani A, Tozzoli R, Brigotti M, Torresani E (2016) Early volume expansion and outcomes of hemolytic uremic syndrome. Pediatrics 137(1). https://doi.org/10.1542/peds.2015-2153
Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, Bremer JP, Brunkhorst R, Busch V, Dengler R, Deuschl G, Fellermann K, Fickenscher H, Gerigk C, Goettsche A, Greeve J, Hafer C, Hagenmuller F, Haller H, Herget-Rosenthal S, Hertenstein B, Hofmann C, Lang M, Kielstein JT, Klostermeier UC, Knobloch J, Kuehbacher M, Kunzendorf U, Lehnert H, Manns MP, Menne TF, Meyer TN, Michael C, Munte T, Neumann-Grutzeck C, Nuernberger J, Pavenstaedt H, Ramazan L, Renders L, Repenthin J, Ries W, Rohr A, Rump LC, Samuelsson O, Sayk F, Schmidt BM, Schnatter S, Schocklmann H, Schreiber S, von Seydewitz CU, Steinhoff J, Stracke S, Suerbaum S, van de Loo A, Vischedyk M, Weissenborn K, Wellhoner P, Wiesner M, Zeissig S, Buning J, Schiffer M, Kuehbacher T, EHEC-HUS consortium, (2012) Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ 345:e4565
Menne J, Delmas Y, Fakhouri F, Licht C, Lommele A, Minetti EE, Provot F, Rondeau E, Sheerin NS, Wang J, Weekers LE, Greenbaum LA (2019) Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol 20:125
Keenswijk W, Raes A, De Clerck M, Vande Walle J (2019) Is plasma exchange efficacious in shiga toxin-associated hemolytic uremic syndrome? A Narrative Review of Current Evidence. Ther Apher Dial 23:118–125
Monet-Didailler C, Chevallier A, Godron-Dubrasquet A, Allard L, Delmas Y, Contin-Bordes C, Brissaud O, Llanas B, Harambat J (2020) Outcome of children with Shiga toxin-associated haemolytic uraemic syndrome treated with eculizumab: a matched cohort study. Nephrol Dial Transplant 35:2147–2153
Walsh PR, Johnson S (2019) Eculizumab in the treatment of Shiga toxin haemolytic uraemic syndrome. Pediatr Nephrol 34:1485–1492
Rosazza C, Cappellari AM, Gandini C, Scola E, Ardissino G (2021) Steroid pulse therapy for severe central nervous system involvement in shiga toxin-producing Escherichia coli-related hemolytic uremic syndrome. Case Rep Pediatr 2021:5587050
Kakoullis L, Papachristodoulou E, Chra P, Panos G (2019) Shiga toxin-induced haemolytic uraemic syndrome and the role of antibiotics: a global overview. J Infect 79:75–94
Giordano M, Baldassarre ME, Palmieri V, Torres DD, Carbone V, Santangelo L, Gentile F, Panza R, Di Mauro F, Capozza M, Di Mauro A, Laforgia N (2019) Management of STEC gastroenteritis: is there a role for probiotics? Int J Environ Res Public Health 16:1649
Timmermans S, van Paassen P (2021) The syndromes of thrombotic microangiopathy: a critical appraisal on complement dysregulation. J Clin Med 10:3034
Sheerin NS, Kavanagh D, Goodship TH, Johnson S (2016) A national specialized service in England for atypical haemolytic uraemic syndrome-the first year’s experience. QJM 109:27–33
Thergaonkar RW, Narang A, Gurjar BS, Tiwari P, Puraswani M, Saini H, Sinha A, Varma B, Mukerji M, Hari P, Bagga A (2018) Targeted exome sequencing in anti-factor H antibody negative HUS reveals multiple variations. Clin Exp Nephrol 22:653–660
Ito S, Hidaka Y, Inoue N, Kaname S, Kato H, Matsumoto M, Miyakawa Y, Mizuno M, Okada H, Shimono A, Matsuda T, Maruyama S, Fujimura Y, Nangaku M, Kagami S (2018) Safety and effectiveness of eculizumab for pediatric patients with atypical hemolytic-uremic syndrome in Japan: interim analysis of post-marketing surveillance. Clin Exp Nephrol 23:112–121
Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C (2017) Haemolytic uraemic syndrome. Lancet 390:681–696
George JN, Nester CM (2014) Syndromes of thrombotic microangiopathy. N Engl J Med 371:1847–1848
Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5:1844–1859
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudailliez B, Bouissou F, Deschenes G, Gie S, Tsimaratos M, Fischbach M, Morin D, Nivet H, Alberti C, Loirat C, French Society of Pediatric Nephrology (2007) Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 18:2392–2400
Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, Pinto S, Goodship TH, Alberti M, Ribes D, Valoti E, Remuzzi G, Noris M, European Working Party on Complement Genetics in Renal Diseases (2013) Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24:475–486
Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, Lommele A, Kupelian V, Gasteyger C, Greenbaum LA, Johnson S, Ogawa M, Licht C, Vande Walle J, Fremeaux-Bacchi V, Global aHUS Registry, (2018) Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int 94:408–418
Thompson RA, Winterborn MH (1981) Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol 46:110–119
Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA (1998) Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 53:836–844
Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, Nester CM, Noris M, Pickering MC, Rodriguez de Cordoba S, Roumenina LT, Sethi S, Smith RJH, Participants C (2017) Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: improving global outcomes” (KDIGO) Controversies Conference. Kidney Int 91:539–551
Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, Burtey S, Niaudet P, Deschenes G, Lebranchu Y, Zuber J, Loirat C (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8:554–562
Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, Mousson C, Mourad G, Bridoux F, Cassuto E, Loirat C, Rondeau E, Delahousse M, Fremeaux-Bacchi V (2013) Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant 13:663–675
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nurnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368:2169–2181
Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, Delmas Y, Douglas K, Furman RR, Gaber OA, Goodship T, Herthelius M, Hourmant M, Legendre CM, Remuzzi G, Sheerin N, Trivelli A, Loirat C (2015) Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 87:1061–1073
Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, Menne J, Minetti EE, Provot F, Rondeau E, Ruggenenti P, Weekers LE, Ogawa M, Bedrosian CL, Legendre CM (2016) Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis 68:84–93
Greenbaum LA, Fila M, Ardissino G, Al-Akash SI, Evans J, Henning P, Lieberman KV, Maringhini S, Pape L, Rees L, van de Kar NC, Vande Walle J, Ogawa M, Bedrosian CL, Licht C (2016) Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int 89:701–711
Palma LM, Langman CB (2016) Critical appraisal of eculizumab for atypical hemolytic uremic syndrome. J Blood Med 7:39–72
Mache CJ, Acham-Roschitz B, Fremeaux-Bacchi V, Kirschfink M, Zipfel PF, Roedl S, Vester U, Ring E (2009) Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 4:1312–1316
Fakhouri F, Fila M, Hummel A, Ribes D, Sellier-Leclerc AL, Ville S, Pouteil-Noble C, Coindre JP, Le Quintrec M, Rondeau E, Boyer O, Provot F, Djeddi D, Hanf W, Delmas Y, Louillet F, Lahoche A, Favre G, Chatelet V, Allain-Launay E, Presne C, Zaloszyc A, Caillard S, Bally S, Raimbourg Q, Tricot L, Mousson C, Le Thuaut A, Loirat C, Fremeaux-Bacchi V (2020) Eculizumab discontinuation in children and adults with atypical haemolytic uremic syndrome: a prospective multicentric study. Blood 137:2438–2449
Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, Miyakawa Y, Ortiz S, Swenson E, Vallee M, Yoon SS, Kavanagh D, Haller H (2020) The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int 97:1287–1296
Ariceta G, Dixon BP, Kim SH, Kapur G, Mauch T, Ortiz S, Vallee M, Denker AE, Kang HG, Greenbaum LA; 312 Study Group (2021) The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int 100:225–237
Scobell RR, Kaplan BS, Copelovitch L (2020) New insights into the pathogenesis of Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol 35:1585–1591
Waters AM, Kerecuk L, Luk D, Haq MR, Fitzpatrick MM, Gilbert RD, Inward C, Jones C, Pichon B, Reid C, Slack MP, Van’t Hoff W, Dillon MJ, Taylor CM, Tullus K (2007) Hemolytic uremic syndrome associated with invasive pneumococcal disease: the United kingdom experience. J Pediatr 151:140–144
Cabrera GR, Fortenberry JD, Warshaw BL, Chambliss CR, Butler JC, Cooperstone BG (1998) Hemolytic uremic syndrome associated with invasive Streptococcus pneumoniae infection. Pediatrics 101:699–703
von Vigier RO, Seibel K, Bianchetti MG (1999) Positive coombs test in pneumococcus-associated hemolytic uremic syndrome. A review of the literature Nephron 82:183–184
Klein PJ, Bulla M, Newman RA, Muller P, Uhlenbruck G, Schaefer HE, Kruger G, Fisher R (1977) Thomsen-Friedenreich antigen in haemolytic-uraemic syndrome. Lancet 2:1024–1025
Ault BH (2000) Factor H and the pathogenesis of renal diseases. Pediatr Nephrol 14:1045–1053
See J, BouMatar R, Baloglu O, Latifi SQ, Talati R, Agarwal HS (2021) Early initiation of eculizumab therapy for Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Blood Cancer 68:e28589
Levandovsky M, Harvey D, Lara P, Wun T (2008) Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): a 24-year clinical experience with 178 patients. J Hematol Oncol 1:23
Bendapudi PK, Hurwitz S, Fry A, Marques MB, Waldo SW, Li A, Sun L, Upadhyay V, Hamdan A, Brunner AM, Gansner JM, Viswanathan S, Kaufman RM, Uhl L, Stowell CP, Dzik WH, Makar RS (2017) Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol 4:e157–e164
Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, Seder RH, Hong SL, Deykin D (1982) Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med 307:1432–1435
Sadler JE, Moake JL, Miyata T, George JN (2004) Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004:407–423
Chung DW, Fujikawa K (2002) Processing of von Willebrand factor by ADAMTS-13. Biochemistry 41:11065–11070
Dong JF (2005) Cleavage of ultra-large von Willebrand factor by ADAMTS-13 under flow conditions. J Thromb Haemost 3:1710–1716
Mancini I, Giacomini E, Pontiggia S, Artoni A, Ferrari B, Pappalardo E, Gualtierotti R, Trisolini SM, Capria S, Facchini L, Codeluppi K, Rinaldi E, Pastore D, Campus S, Caria C, Caddori A, Nicolosi D, Giuffrida G, Agostini V, Roncarati U, Mannarella C, Fragasso A, Podda GM, Birocchi S, Cerbone AM, Tufano A, Menna G, Pizzuti M, Ronchi M, De Fanti A, Amarri S, Defina M, Bocchia M, Ceru S, Gattillo S, Rosendaal FR, Peyvandi F (2020) The HLA variant rs6903608 is associated with disease onset and relapse of immune-mediated thrombotic thrombocytopenic purpura in caucasians. J Clin Med 9:3379
Bell WR, Braine HG, Ness PM, Kickler TS (1991) Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med 325:398–403
Padmanabhan A, Connelly-Smith L, Aqui N, Balogun RA, Klingel R, Meyer E, Pham HP, Schneiderman J, Witt V, Wu Y, Zantek ND, Dunbar NM, Schwartz GEJ (2019) Guidelines on the use of therapeutic apheresis in clinical practice—evidence-based approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J Clin Apher 34:171–354
Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA (1991) Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 325:393–397
Kremer Hovinga JA, Coppo P, Lammle B, Moake JL, Miyata T, Vanhoorelbeke K (2017) Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 3:17020
Biesert L, Suhartono H (1998) Solvent/detergent treatment of human plasma—a very robust method for virus inactivation. Validated virus safety of OCTAPLAS. Vox Sang 74(Suppl 1):207–212
Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knobl P, Wu H, Artoni A, Westwood JP, Mansouri Taleghani M, Jilma B, Callewaert F, Ulrichts H, Duby C, Tersago D, TITAN Investigators (2016) Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 374:511–522
Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, Biswas D, De Winter H, Zeldin RK, HERCULES Investigators (2019) Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med 380:335–346
Hassenpflug WA, Obser T, Bode J, Oyen F, Budde U, Schneppenheim S, Schneppenheim R, Brehm MA (2018) Genetic and functional characterization of ADAMTS13 variants in a patient cohort with Upshaw-Schulman syndrome investigated in Germany. Thromb Haemost 118:709–722
Kremer Hovinga JA, Braschler TR, Buchkremer F, Farese S, Hengartner H, Lovey PY, Largiader CR, Mansouri Taleghani B, Tarasco E (2020) Insights from the hereditary thrombotic thrombocytopenic purpura registry: discussion of key findings based on individual cases from Switzerland. Hamostaseologie 40:S5–S14
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, Coppo R, Emma F, Johnson S, Karpman D, Landau D, Langman CB, Lapeyraque AL, Licht C, Nester C, Pecoraro C, Riedl M, van de Kar NC, Van de Walle J, Vivarelli M, Fremeaux-Bacchi V, HUS International (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31:15–39
Vaisbich MH, Braga A, Gabrielle M, Bueno C, Piazzon F, Kok F (2017) Thrombotic microangiopathy caused by methionine synthase deficiency: diagnosis and treatment pitfalls. Pediatr Nephrol 32:1089–1092
Cornec-Le Gall E, Delmas Y, De Parscau L, Doucet L, Ogier H, Benoist JF, Fremeaux-Bacchi V, Le Meur Y (2014) Adult-onset eculizumab-resistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am J Kidney Dis 63:119–123
Grange S, Bekri S, Artaud-Macari E, Francois A, Girault C, Poitou AL, Benhamou Y, Vianey-Saban C, Benoist JF, Chatelet V, Tamion F, Guerrot D (2015) Adult-onset renal thrombotic microangiopathy and pulmonary arterial hypertension in cobalamin C deficiency. Lancet 386:1011–1012
Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, Ji W, Overton JD, Mane SM, Nurnberg G, Altmuller J, Thiele H, Morin D, Deschenes G, Baudouin V, Llanas B, Collard L, Majid MA, Simkova E, Nurnberg P, Rioux-Leclerc N, Moeckel GW, Gubler MC, Hwa J, Loirat C, Lifton RP (2013) Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45:531–536
Bezdicka M, Pavlicek P, Blahova K, Hacek J, Zieg J (2020) Various phenotypes of disease associated with mutated DGKE gene. Eur J Med Genet 63:103953
Quaggin SE (2013) DGKE and atypical HUS. Nat Genet 45:475–476
Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, Grinyo JM, Macia M, Mendizabal S, Praga M, Roman E, Torra R, Valdes F, Vilalta R, Rodriguez de Cordoba S (2015) An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document Nefrologia 35:421–447
Epperla N, Li A, Logan B, Fretham C, Chhabra S, Aljurf M, Chee L, Copelan E, Freytes CO, Hematti P, Lazarus HM, Litzow M, Nishihori T, Olsson RF, Prestidge T, Saber W, Wirk B, Yared JA, Loren A, Pasquini M (2020) Incidence, risk factors for and outcomes of transplant-associated thrombotic microangiopathy. Br J Haematol 189:1171–1181
Jodele S, Dandoy CE, Lane A, Laskin BL, Teusink-Cross A, Myers KC, Wallace G, Nelson A, Bleesing J, Chima RS, Hirsch R, Ryan TD, Benoit S, Mizuno K, Warren M, Davies SM (2020) Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood 135:1049–1057
Acknowledgements
SS would like to thank the Division of Anatomic Pathology, Innovation fund, Mayo Clinic.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
LMPP is a speaker for Alexion Brazil, and scientific consultant for Apellis and Novartis.
Disclosure
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Palma, L.M.P., Vaisbich-Guimarães, M.H., Sridharan, M. et al. Thrombotic microangiopathy in children. Pediatr Nephrol 37, 1967–1980 (2022). https://doi.org/10.1007/s00467-021-05370-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-05370-8