
Abstract
Background
Hemolytic uremic syndrome caused by invasive pneumococcal disease (P-HUS) is rare in children and adolescents, but accompanied by high mortality in the acute phase and complicated by long-term renal sequelae. Abnormalities in the alternative complement pathway may additionally be contributing to the course of the disease but also to putative treatment options.
Methods
Retrospective study to assess clinical course and laboratory data of the acute phase and outcome of children with P-HUS.
Results
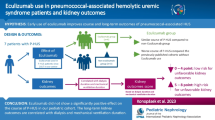
We report on seven children (median age 12 months, range 3–28 months) diagnosed with P-HUS. Primary organ manifestation was meningitis in four and pneumonia in three patients. All patients required dialysis which could be discontinued in five of them after a median of 25 days. In two patients, broad functional and genetic complement analysis was performed and revealed alternative pathway activation and risk haplotypes in both. Three patients were treated with the complement C5 inhibitor eculizumab. During a median follow-up time of 11.3 years, one patient died due to infectious complications after transplantation. Two patients showed no signs of renal sequelae.
Conclusions
Although pathophysiology in P-HUS remains as yet incompletely understood, disordered complement regulation seems to provide a clue to additional insights for pathology, diagnosis, and even targeted treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemolytic uremic syndrome (HUS) is the most common cause of acute intrarenal kidney injury in childhood, mainly caused by Shigatoxin-producing Escherichia coli (STEC-HUS) [1]. Non-STEC-HUS can be caused by anomalies in complement regulating genes or by acquired, secondary alternative complement pathway activation. One peculiar form of HUS is accompanied by invasive infections with Streptococcus pneumoniae (invasive pneumococcal disease, IPD). Although pneumococcal HUS (P-HUS) represents approximately 5–15% of all HUS cases [2], robust data about its clinical course, treatment, and outcome are lacking. New insights into disease pathophysiology of P-HUS suggesting a potential role of alternative complement pathway activation contributing to disease development have been reported [2], but clinical reports about complement activation in P-HUS are still rare.
Here, we provide a case series of P-HUS underlining the potential role of complement activation and, hereby, a potential treatment option.
Patients and methods
We retrospectively analyzed clinical and laboratory data of the acute phase in children diagnosed with P-HUS between the years 1996 and 2019. Diagnosis of P-HUS, was based on recent recommendations including evidence for HUS (acute kidney injury, microangiopathic hemolytic anemia with schistocytes, thrombocytopenia), evidence of IPD (positive culture of blood or another sterile biological fluid), and absence of disseminated intravasal coagulation [2].
We investigated demographic (gender, age, body height and weight), microbiological (primary site of infection, results of bacterial cultures and serology), laboratory data (hemoglobin, platelet count, serum creatinine, Coombs test, complement diagnostics, when available, including genetics analyzing the following genes: ADAMTS13, C3, CFB, CFH, CFHR-1-5, CFI, DGKE, MCP/CD46, MMACHC, and THMBD) and clinical data. Further, the duration and modality of kidney replacement therapy was encountered as well as administration of the complement C5 inhibitor eculizumab. Follow-up parameters were analyzed including estimated GFR (eGFR) using the Schwartz formula [3], arterial hypertension (defined as blood pressure values above the 95th percentile in at least three individual measurements [4]), proteinuria (defined by the protein-creatinine ratio (g/g) in spot urine as absence of proteinuria (< 0.2 or negative) or positive proteinuria (> 0.2)), and the occurrence of relapses until the last documented visit.
Results
Between 1996 and 2019, seven patients with definite diagnosis of P-HUS were treated at our hospital. Clinical presentation and outcome are listed in Table 1. Median patient’s age at presentation was 12 months [range 3–28 months].
Acute phase: All patients showed acute kidney injury, hemolytic anemia with fragmentocytes, and thrombocytopenia within the acute phase of disease. Two patients had a positive Coombs test. IPD was present in all patients, with primary manifestation of meningitis in four patients and pneumonia in three patients. Despite vaccination with pneumococcal conjugate vaccine (PCV13), serotype S3 (which is included in PCV13) was detected in two patients, and serotype S15A was detected in one patient. Functional and genetic complement analyses were performed in two cases, revealing alternative pathway activation and a risk haplotype in the complement regulator membrane cofactor protein (MCP-H2, GGAAC, heterozygote mutation), but no other genetic anomalies in complement-regulating genes.
All patients needed kidney replacement therapy during the acute phase, which was performed as peritoneal dialysis. While five patients recovered during a median time on dialysis of 25 days [range 20–42], two patients developed stage 5 chronic kidney disease (CKD 5).
Three patients (patients 5, 6 and 7) were treated with the complement C5 inhibitor eculizumab due to initial signs of complement activation and/or supposed complement-mediated HUS. Patient no. 5 showed low serum C3 level. Further complement diagnostics were not performed. In this patient, eculizumab treatment was stopped after one dose due to severe infection and missing data about its effectivity in the year 2014. Patient no. 6 received eculizumab because of activated alternative complement pathway. Three days after first administration, thrombocyte count increased (from 32 to 45 thrombocytes/nl without transfusion) and LDH activity decreased (from 3373 to 1411 U/l) markedly. In the absence of known genetic anomalies in complement-regulating genes, eculizumab was discontinued 6 months after recovery. Patient no. 7 received eculizumab and showed rapid recovery similarly to patient no. 6 (thrombocytes from 66 to 183/nl and LDH activity from 4384 to 1908 U/l within two days). Eculizumab was discontinued after three doses as kidney function improved, and no genetic anomalies were detected.
Follow-up: During a median follow-up time of 11 years and 4 months (range 29 months–17 years, 4months), none of the patients experienced a relapse of P-HUS. Both patients with CKD 5 received a kidney transplant during the follow-up. One patient deceased due to infectious complications after transplantation. Three patients exhibited renal sequelae with reduced eGFR (n = 3), arterial hypertension (n = 3), and proteinuria (n = 2). Two patients recovered (eGFR > 90 ml/min/1.73 m2).
Discussion
Within this study, we report about the clinical course and outcome of seven children with HUS, associated with IPD within a time period of 24 years. Despite national vaccination recommendations, IPD remains a major cause of morbidity and mortality in children living in Germany [5]. As reporting IPD is voluntary, exact data about the incidence of IPD are not available. Young children with a median age of 13 months are known to have the highest risk of IPD [5]. Hence, it is not surprising that the prevalence of P-HUS is highest in children under 2 years of age. Accordingly, the median age in our cohort was 12 months.
As reported previously, 92 patients with HUS were managed at our hospital between 1996 and 2004 [6], with evidence of P-HUS in five of them (5.4%). Therefore, the estimated rate of P-HUS recorded in our hospital (5.4%) is lower compared with the rate reported in the literature, implicating the need for resilient and reproducible diagnostic criteria [7, 8]. In contrast to other reports [9], we observed a higher proportion of meningitis compared with pneumonia. The finding of serotype S3 in two children despite having received appropriate vaccination (PCV13) underlines the potential risk of vaccination failure.
As discussed in a recent review article [2], the pathophysiology of P-HUS remains incompletely understood. The mechanism of endothelial damage by exposure of Thomsen-Friedenreich antigen, revealed by S. pneumoniae neuraminidase activity, remains widely accepted [2]. However, the subsequent binding of preformed IgM antibodies [10], resulting in a positive direct Coombs test [11], is more and more challenged by contradictory findings [2] and cannot be confirmed in our cohort with only two patients presenting with a positive Coombs test. Interestingly, alternative complement pathway activation might play a role in determining why some patients with IPD develop P-HUS while others do not. As a result of neuraminidase desialylation, factor H binding to C3 convertase decreases, resulting in alternative complement activation in vitro [12]. Moreover, there are reports on alternative complement pathway activation in vivo and partially pathogenic variants in complement regulating genes [13,14,15]. Our findings support an involvement of the alternative complement system in P-HUS pathology. Both patients who received comprehensive complement analysis showed significant activation of the alternative pathway during the acute phase. Genetic analysis revealed no definite pathogenic variants in complement-regulating genes. Nevertheless, the MCP-H2 risk haplotype was found in both patients, reported to be associated with a 2–3-fold increased risk for developing aHUS and contributing to its pathophysiology in the context of the multiple-hits theory [16]. Three of our patients were treated with eculizumab. Eculizumab has been described in two patients with P-HUS previously [14, 17] — both showed rapid hematological recovery, and kidney replacement therapy could be discontinued.
P-HUS is associated with a high morbidity and mortality, indicated by 10.1% of patients developing CKD 5, 16% developing CKD and hypertension, and 12.3% deaths within the acute phase, especially in those cases with meningitis as primary manifestation [18]. In our cohort, no patient died during the acute disease course, and dialysis could be discontinued in five patients.
During the median follow-up time of 11 years and 4 months, two patients developed CKD 5 and three patients showed renal sequelae, while two patients showed recovery of kidney function. Of note, patient no. 6 who was treated for the longest period with eculizumab showed the most favorable outcome despite showing the most severe complement activation.
These findings indicate that besides other factors, the terminal pathway activation is critical for eculizumab to be effective in treatment of patients with P-HUS. Although complement activation data are lacking in five of seven patients, and accordingly, this limits the evidence of the study, our findings support the hypothesis of complement activation in P-HUS and favor the potential benefit of complement inhibition in selected patients.
References
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, Coppo R, Emma F, Johnson S, Karpman D, Landau D, Langman CB, Lapeyraque AL, Licht C, Nester C, Pecoraro C, Riedl M, van de Kar NC, Van de Walle J, Vivarelli M, Fremeaux-Bacchi V, HUS International (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31:15–39
Scobell RR, Kaplan BS, Copelovitch L (2020) New insights into the pathogenesis of Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol 35:1585–1591
Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, Furth SL (2009) New equations to estimate GFR in children with CKD. J Am Soc Nephrol 20:629–637
Lurbe E, Agabiti-Rosei E, Cruickshank JK, Dominiczak A, Erdine S, Hirth A, Invitti C, Litwin M, Mancia G, Pall D, Rascher W, Redon J, Schaefer F, Seeman T, Sinha M, Stabouli S, Webb NJ, Wuhl E, Zanchetti A (2016) 2016 European Society of Hypertension guidelines for the management of high blood pressure in children and adolescents. J Hypertens 34:1887–1920
Perniciaro S, Imohl M, Fitzner C, van der Linden M (2019) Regional variations in serotype distribution and vaccination status in children under six years of age with invasive pneumococcal disease in Germany. PLoS One 14:e0210278
Vaterodt L, Holle J, Huseman D, Muller D, Thumfart J (2018) Short- and long-term renal outcome of hemolytic-uremic syndrome in childhood. Front Pediatr 6:220
Lawrence J, Gwee A, Quinlan C (2018) Pneumococcal haemolytic uraemic syndrome in the postvaccine era. Arch Dis Child 103:957–961
Bender JM, Ampofo K, Byington CL, Grinsell M, Korgenski K, Daly JA, Mason EO, Pavia AT (2010) Epidemiology of Streptococcus pneumoniae-induced hemolytic uremic syndrome in Utah children. Pediatr Infect Dis J 29:712–716
Waters AM, Kerecuk L, Luk D, Haq MR, Fitzpatrick MM, Gilbert RD, Inward C, Jones C, Pichon B, Reid C, Slack MP, Van't Hoff W, Dillon MJ, Taylor CM, Tullus K (2007) Hemolytic uremic syndrome associated with invasive pneumococcal disease: the United kingdom experience. J Pediatr 151:140–144
Smith A, Johnston C, Inverarity D, Slack M, Paterson GK, Diggle M, Mitchell T (2013) Investigating the role of pneumococcal neuraminidase A activity in isolates from pneumococcal haemolytic uraemic syndrome. J Med Microbiol 62:1735–1742
von Vigier RO, Fossali E, Crosazzo L, Bianchetti MG (2005) Positive Coombs test in postpneumococcal hemolytic-uremic syndrome. Pediatr Infect Dis J 24:1028–1029
Kerr H, Wong E, Makou E, Yang Y, Marchbank K, Kavanagh D, Richards A, Herbert AP, Barlow PN (2017) Disease-linked mutations in factor H reveal pivotal role of cofactor activity in self-surface-selective regulation of complement activation. J Biol Chem 292:13345–13360
Szilagyi A, Kiss N, Bereczki C, Talosi G, Racz K, Turi S, Gyorke Z, Simon E, Horvath E, Kelen K, Reusz GS, Szabo AJ, Tulassay T, Prohaszka Z (2013) The role of complement in Streptococcus pneumoniae-associated haemolytic uraemic syndrome. Nephrol Dial Transplant 28:2237–2245
Gilbert RD, Nagra A, Haq MR (2013) Does dysregulated complement activation contribute to haemolytic uraemic syndrome secondary to Streptococcus pneumoniae? Med Hypotheses 81:400–403
Bitzan M, AlKandari O, Whittemore B, Yin XL (2018) Complement depletion and Coombs positivity in pneumococcal hemolytic uremic syndrome (pnHUS). Case series and plea to revisit an old pathogenetic concept. Int J Med Microbiol 308:1096–1104
Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grunfeld JP, Niaudet P, Lesavre P, Fremeaux-Bacchi V (2012) Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 82:454–464
Jeantet G, Pernin V, Brunot V, Roccabianca A, Macombe A, Szwarc I, Klouche K, Loirat C, Mourad G, Fremeaux-Bacchi V, Le Quintrec M (2019) Successful treatment of a Streptococcus pneumoniae-associated haemolytic uraemic syndrome by eculizumab. Clin Kidney J 12:106–109
Copelovitch L, Kaplan BS (2008) Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Nephrol 23:1951–1956
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from all individual participants included in the study
Conflict of interest
None declared
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holle, J., Habbig, S., Gratopp, A. et al. Complement activation in children with Streptococcus pneumoniae associated hemolytic uremic syndrome. Pediatr Nephrol 36, 1311–1315 (2021). https://doi.org/10.1007/s00467-021-04952-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-04952-w