
Abstract
Background
Copy number variants (CNVs) are increasingly recognized as an important cause of congenital malformations and likely explain over 16 % of cases of congenital anomalies of the kidney and urinary tract (CAKUT). Here, we illustrate how a molecular diagnosis of CNV can be beneficial to the clinical management of a pediatric patient presenting with CAKUT and other organ defects.
Methods
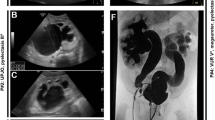
We describe a 14-year-old girl with a large de novo deletion of chromosome 3q13.31-22.1 that disrupts 101 known genes. The patient presented with CAKUT, neurodevelopmental delay, agenesis of corpus callosum (ACC), cardiac malformations, electrolyte and endocrine disorders, skeletal abnormalities and dysmorphic features. We performed extensive annotation of the deleted region to prioritize genes for specific phenotypes and to predict future disease risk.
Results
Our case defined new minimal chromosomal candidate regions for both CAKUT and ACC. The presence of the CASR gene in the deleted interval predicted a diagnosis of hypocalciuric hypercalcemia, which was confirmed by the serum and urine chemistries. Our gene annotation explained clinical hypothyroidism and predicted that the index case is at increased risk of thoracic aortic aneurysm, renal cell carcinoma and myeloproliferative disorder.
Conclusions
Extended annotation of CNV regions refines the diagnosis and uncovers previously unrecognized phenotypic features. This approach enables personalized treatment and prevention strategies in patients harboring genomic deletions.




Similar content being viewed by others

References
Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, Nees SN, Verbitsky M, Perry BJ, Sterken R, Lozanovski VJ, Materna-Kiryluk A, Barlassina C, Kini A, Corbani V, Carrea A, Somenzi D, Murtas C, Ristoska-Bojkovska N, Izzi C, Bianco B, Zaniew M, Flogelova H, Weng PL, Kacak N, Giberti S, Gigante M, Arapovic A, Drnasin K, Caridi G, Curioni S, Allegri F, Ammenti A, Ferretti S, Goj V, Bernardo L, Jobanputra V, Chung WK, Lifton RP, Sanders S, State M, Clark LN, Saraga M, Padmanabhan S, Dominiczak AF, Foroud T, Gesualdo L, Gucev Z, Allegri L, Latos-Bielenska A, Cusi D, Scolari F, Tasic V, Hakonarson H, Ghiggeri GM, Gharavi AG (2012) Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91:987–997
Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, Nezarati MM, Asamoah A, Jackson KE, Gowans GC, Martin JA, Carmany EP, Stockton DW, Schnur RE, Penney LS, Martin DM, Raskin S, Leppig K, Thiese H, Smith R, Aberg E, Niyazov DM, Escobar LF, El-Khechen D, Johnson KD, Lebel RR, Siefkas K, Ball S, Shur N, McGuire M, Brasington CK, Spence JE, Martin LS, Clericuzio C, Ballif BC, Shaffer LG, Eichler EE (2012) Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med 367:1321–1331
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE (2011) A copy number variation morbidity map of developmental delay. Nat Genet 43:838–846
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J (2008) Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320:539–543
Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, GROUP, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K (2008) Large recurrent microdeletions associated with schizophrenia. Nature 455:232–236
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M (2007) Strong association of de novo copy number mutations with autism. Science 316:445–449
Soemedi R, Wilson IJ, Bentham J, Darlay R, Topf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue GM, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman TJ, Hall D, Rauch A, Devriendt K, Gewillig M, O’Sullivan J, Winlaw DS, Bu’Lock F, Brook JD, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD (2012) Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 91:489–501
Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, Lionel AC, D’Amours G, Lemyre E, Cullum R, Bigras JL, Thibeault M, Chetaille P, Montpetit A, Khairy P, Overduin B, Klaassen S, Hoodless P, Nemer M, Stewart AF, Boerkoel C, Scherer SW, Richter A, Dube MP, Andelfinger G (2012) Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet 8:e1002903
Dauber A, Yu Y, Turchin MC, Chiang CW, Meng YA, Demerath EW, Patel SR, Rich SS, Rotter JI, Schreiner PJ, Wilson JG, Shen Y, Wu BL, Hirschhorn JN (2011) Genome-wide association of copy-number variation reveals an association between short stature and the presence of low-frequency genomic deletions. Am J Hum Genet 89:751–759
Zahnleiter D, Uebe S, Ekici AB, Hoyer J, Wiesener A, Wieczorek D, Kunstmann E, Reis A, Doerr HG, Rauch A, Thiel CT (2013) Rare copy number variants are a common cause of short stature. PLoS Genet 9:e1003365
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C (2011) American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13:680–685
Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, Tommerup N, Eichler EE, Bellanne-Chantelot C (2007) Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81:1057–1069
Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, Clauin S, Deschenes G, Bouissou F, Bensman A, Bellanne-Chantelot C (2006) Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 17:497–503
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M (2007) PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17:1665–1674
Huang N, Lee I, Marcotte EM, Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154
Mackie Ogilvie C, Rooney SC, Hodgson SV, Berry AC (1998) Deletion of chromosome 3q proximal region gives rise to a variable phenotype. Clin Genet 53:220–222
Okada N, Hasegawa T, Osawa M, Fukuyama Y (1987) A case of de novo interstitial deletion 3q. J Med Genet 24:305–308
Jenkins MB, Stang HJ, Davis E, Boyd L (1985) Deletion of the proximal long arm of chromosome 3 in an infant with features of Turner syndrome. Ann Genet 28:42–44
Genuardi M, Calvieri F, Tozzi C, Coslovi R, Neri G (1994) A new case of interstitial deletion of chromosome 3q, del(3q)(q13.12q21.3), with agenesis of the corpus callosum. Clin Dysmorphol 3:292–296
McMorrow LE, Reid CS, Coleman J, Medeiros A, D’Andrea M, Santucci T, McCormack MK (1986) A new interstitial deletion of the long arm of chromosome 3. Am J Hum Genet 39:A124
Fujita H, Meng J, Kawamura M, Tozuka N, Ishii F, Tanaka N (1992) Boy with a chromosome del (3)(q12q23) and blepharophimosis syndrome. Am J Med Genet 44:434–436
Lawson-Yuen A, Berend SA, Soul JS, Irons M (2006) Patient with novel interstitial deletion of chromosome 3q13.1q13.3 and agenesis of the corpus callosum. Clin Dysmorphol 15:217–220
Molin AM, Andrieux J, Koolen DA, Malan V, Carella M, Colleaux L, Cormier-Daire V, David A, de Leeuw N, Delobel B, Duban-Bedu B, Fischetto R, Flinter F, Kjaergaard S, Kok F, Krepischi AC, Le Caignec C, Ogilvie CM, Maia S, Mathieu-Dramard M, Munnich A, Palumbo O, Papadia F, Pfundt R, Reardon W, Receveur A, Rio M, Ronsbro Darling L, Rosenberg C, Sa J, Vallee L, Vincent-Delorme C, Zelante L, Bondeson ML, Anneren G (2012) A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet 49:104–109
Jiang H, Guo W, Liang X, Rao Y (2005) Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120:123–135
Liu KJ, Arron JR, Stankunas K, Crabtree GR, Longaker MT (2007) Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature 446:79–82
Rosenfeld JA, Ballif BC, Martin DM, Aylsworth AS, Bejjani BA, Torchia BS, Shaffer LG (2010) Clinical characterization of individuals with deletions of genes in holoprosencephaly pathways by aCGH refines the phenotypic spectrum of HPE. Hum Genet 127:421–440
Eccles MR, Schimmenti LA (1999) Renal-coloboma syndrome: a multi-system developmental disorder caused by PAX2 mutations. Clin Genet 56:1–9
Deng FM, Liang FX, Tu L, Resing KA, Hu P, Supino M, Hu CC, Zhou G, Ding M, Kreibich G, Sun TT (2002) Uroplakin IIIb, a urothelial differentiation marker, dimerizes with uroplakin Ib as an early step of urothelial plaque assembly. J Cell Biol 159:685–694
Hu P, Deng FM, Liang FX, Hu CM, Auerbach AB, Shapiro E, Wu XR, Kachar B, Sun TT (2000) Ablation of uroplakin III gene results in small urothelial plaques, urothelial leakage, and vesicoureteral reflux. J Cell Biol 151:961–972
Jenkins D, Bitner-Glindzicz M, Malcolm S, Hu CC, Allison J, Winyard PJ, Gullett AM, Thomas DF, Belk RA, Feather SA, Sun TT, Woolf AS (2005) De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J Am Soc Nephrol 16:2141–2149
Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, Utsch B, Sayer JA, Lillo C, Jimeno D, Coucke P, De Paepe A, Reinhardt R, Klages S, Tsuda M, Kawakami I, Kusakabe T, Omran H, Imm A, Tippens M, Raymond PA, Hill J, Beales P, He S, Kispert A, Margolis B, Williams DS, Swaroop A, Hildebrandt F (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 37:282–288
Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW (2010) Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 86:949–956
Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, Branney PA, Fisher M, Lee GJ, Simpson MA, He Y, Bradshaw TY, Blaumeiser B, Winship WS, Reardon W, Maher ER, FitzPatrick DR, Wuyts W, Zenker M, Lamarche-Vane N, Trembath RC (2011) Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet 88:574–585
Niimi H, Inomata H, Sasaki N, Nakajima H (1982) Congenital isolated thyrotrophin releasing hormone deficiency. Arch Dis Child 57:877–878
Collu R, Tang J, Castagne J, Lagace G, Masson N, Huot C, Deal C, Delvin E, Faccenda E, Eidne KA, Van Vliet G (1997) A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab 82:1561–1565
Persani L, Ferretti E, Borgato S, Faglia G, Beck-Peccoz P (2000) Circulating thyrotropin bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab 85:3631–3635
Beck-Peccoz P, Amr S, Menezes-Ferreira MM, Faglia G, Weintraub BD (1985) Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism. Effect of treatment with thyrotropin-releasing hormone. N Engl J Med 312:1085–1090
Miura Y, Perkel VS, Papenberg KA, Johnson MJ, Magner JA (1989) Concanavalin-A, lentil, and ricin lectin affinity binding characteristics of human thyrotropin: differences in the sialylation of thyrotropin in sera of euthyroid, primary, and central hypothyroid patients. J Clin Endocrinol Metab 69:985–995
Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, Park J, Hebert SC, Seidman CE, Seidman JG (1994) Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet 8:303–307
Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG (1993) Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75:1297–1303
Pollak MR, Chou YH, Marx SJ, Steinmann B, Cole DE, Brandi ML, Papapoulos SE, Menko FH, Hendy GN, Brown EM (1994) Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J Clin Invest 93:1108–1112
Chen YZ, Matsushita MM, Robertson P, Rieder M, Girirajan S, Antonacci F, Lipe H, Eichler EE, Nickerson DA, Bird TD, Raskind WH (2012) Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 69:630–635
Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM (2010) Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 87:701–707
Bodmer D, Eleveld M, Kater-Baats E, Janssen I, Janssen B, Weterman M, Schoenmakers E, Nickerson M, Linehan M, Zbar B, van Kessel AG (2002) Disruption of a novel MFS transporter gene, DIRC2, by a familial renal cell carcinoma-associated t(2;3)(q35;q21). Hum Mol Genet 11:641–649
Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, Babic M, Lin M, Carmagnac A, Lee YK, Kok CH, Gagliardi L, Friend KL, Ekert PG, Butcher CM, Brown AL, Lewis ID, To LB, Timms AE, Storek J, Moore S, Altree M, Escher R, Bardy PG, Suthers GK, D’Andrea RJ, Horwitz MS, Scott HS (2011) Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 43:1012–1017
Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, Dafou D, Kilo T, Smithson S, Lunt P, Murday VA, Hodgson S, Keenan R, Pilz DT, Martinez-Corral I, Makinen T, Mortimer PS, Jeffery S, Trembath RC, Mansour S (2011) Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet 43:929–931
Malan V, Chevallier S, Soler G, Coubes C, Lacombe D, Pasquier L, Soulier J, Morichon-Delvallez N, Turleau C, Munnich A, Romana S, Vekemans M, Cormier-Daire V, Colleaux L (2010) Array-based comparative genomic hybridization identifies a high frequency of copy number variations in patients with syndromic overgrowth. Eur J Hum Genet 18:227–232
Lawson CT, Toomes C, Fryer A, Carette MJ, Taylor GM, Fukushima Y, Dixon MJ (1995) Definition of the blepharophimosis, ptosis, epicanthus inversus syndrome critical region at chromosome 3q23 based on the analysis of chromosomal anomalies. Hum Mol Genet 4:963–967
Wolstenholme J, Brown J, Masters KG, Wright C, English CJ (1994) Blepharophimosis sequence and diaphragmatic hernia associated with interstitial deletion of chromosome 3 (46, XY, del(3)(q21q23)). J Med Genet 31:647–648
Jewett T, Rao PN, Weaver RG, Stewart W, Thomas IT, Pettenati MJ (1993) Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) associated with interstitial deletion of band 3q22: review and gene assignment to the interface of band 3q22.3 and 3q23. Am J Med Genet 47:1147–1150
Acknowledgments
We are grateful to the devoted family of the index case for their ongoing participation. This study is the result of scientific collaboration between the Department of Medical Genetics, Poznan University of Medical Sciences in Poland and the Division of Nephrology at Columbia University in New York, USA. Our funding sources include: A.M.K., A.L.B., and The Polish Registry of Congenital Malformations (PRCM) are supported by the Polish Ministry of Health; A.G.G. and K.K. are supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01DK080099 and K23-DK090207, respectively. S.S.C. is supported by the American Heart Association Grant in Aid 13GRNT14680075 and by the American Society of Nephrology Carl W Gottschalk Research Scholar Grant.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Anna Materna-Kiryluk and Krzysztof Kiryluk contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic Supplementary Material Dataset 1
Reference genes in the minimal candidate regions for CAKUT and ACC. Column legend: chromosome, transcript start (bp, hg-18), transcript stop (bp, hg-18), gene name, predicted probability of haploinsufficiency according to the probabilistic model of Huang et al. [15], membership in the minimal candidate region for CAKUT and ACC (TXT 3 kb)
Rights and permissions
About this article
Cite this article
Materna-Kiryluk, A., Kiryluk, K., Burgess, K.E. et al. The emerging role of genomics in the diagnosis and workup of congenital urinary tract defects: a novel deletion syndrome on chromosome 3q13.31-22.1. Pediatr Nephrol 29, 257–267 (2014). https://doi.org/10.1007/s00467-013-2625-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-013-2625-2