
Abstract
Nephropathic cystinosis (NC) is an autosomal recessive disorder caused by mutations of the CTNS gene that encodes for a cystine transmembrane transporter. Several mutations have been described in the coding and promoter regions of the CTNS gene in affected individuals. We selected three patients with NC from two unrelated families, in whom sequence analysis of the CTNS gene detected only one or no mutations. Total RNA was isolated from peripheral blood mononuclear cells or fibroblasts and CTNS transcripts were analyzed. We observed a skipping of exon 5 (85 bp) in two siblings and an intron 9 retention of 75 bp associated with partial replication of exon 9 in the third patient. Genomic DNA analysis of intron regions surrounding exon 5 showed a point mutation in the hypothetical lariat branch site of intron 4 at position –24 (c.141–24 T > C) in the first two patients and a duplication of 266 bp including a part of exon and intron 9 in the third patient. Analysis of CTNS gene transcripts allowed identification of mutations in patients in whom CTNS mutations could not be detected by traditional DNA sequencing. These results support the hypothesis that cystinosis is a monogenic disorder.
Similar content being viewed by others


Introduction
Nephropathic cystinosis (NC) (OMIN 219800) is a rare autosomal recessive disorder (incidence in 1:100,000–200,000 live births), which presents with renal proximal tubular dysfunction (Fanconi syndrome) during infancy and progresses to end-stage renal failure during the first decade of life if not treated with cysteamine [1]. Over time, patients with NC also present with dysfunction of several organs, including eyes, thyroid, liver, pancreas, male gonad, muscles, and brain [2]. NC is caused by mutations in the CTNS gene [3]. It encodes for cystinosin, an amino-acid-cystine carrier that is predicted to have seven transmembrane domains and is primarily expressed in the lysosomal membrane [4]. Lack of cystinosin activity causes cystine accumulation and results in formation of intralysosomal cystine crystals. Approximately 10–20% of CTNS transcripts encode for a second cystinosin isoform, termed cystinosin-LKG, which results from an alternative splicing of exon 12 and is expressed in the plasma membrane and other cytosolic vesicles, in addition to lysosomes [5].
The CTNS gene maps to chromosome 17p13 and is composed of 12 exons, with the start codon located on exon 3 [3]. The most common CTNS mutation in northern Europe is a 57-kb deletion that originated in Germany around AD 500 [1]. To date, more than 90 mutations have been described in the coding region or flanking intronic regions of the gene, as well as in the CTNS promoter, in patients with cystinosis. Of these, approximately 70 mutations cause NC. These include small deletions or insertions, missense or nonsense mutations, and splicing mutations [3, 6–13].
In clinical practice, the diagnosis of NC is sustained by the demonstration of increased intraleucocyte cystine levels. Genetic tests have primarily a confirmatory value and allow for genetic counseling. Evidence suggests that NC is a monogenic disease, although the mutation detection rate is not 100%. Shotelersuk et al., for example, sequenced the CTNS gene in 108 American patients with NC and failed to detect mutations in 19% of affected individuals [6]. The CTNS promoter was, however, not analyzed in their study. Similar studies that also included the promoter region showed only heterozygous or no mutations in 18% of 46 Italian patients and in 6% of 108 French patients [9, 11].
In this work, we studies CTNS gene transcripts in three patients with NC from two families to search for mutations that were not detected at the genomic level.
Patients and methods
Patients
Among 49 Dutch patients with NC from 44 different families, we selected three patients in whom sequence analysis failed to show homozygous or double heterozygous mutations in the CTNS gene (coding regions, intron/exon boundaries, or promoter region). Clinical data of these patients are summarized in Table 1. The diagnosis of NC was established in all patients by demonstration of increased intraleucocyte cystine levels and corneal cystine crystal depositions. Informed consent was obtained to perform the additional genetic studies included in this work.
Peripheral blood mononuclear cell isolation and fibroblasts cell lines
Anticoagulated whole blood samples were diluted with phosphate-buffered saline (PBS) (1:1), layered over Ficoll Histopaque 1077 (Sigma-Aldrich, St. Louis, MO, USA), and centrifuged at 1,800 g for 20 min at room temperature. The peripheral blood mononuclear cell (PBMC) layer was removed and washed twice with PBS following two centrifugation steps at 250 g for 10 min. RNA studies were performed in patient 3 on a previously established fibroblast cell line from that patient [14].
Reverse transcriptase–polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from PBMCs or fibroblasts using TRIzol® reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). RNA was reverse-transcribed into complementary DNA (cDNA) using a first-strand cDNA synthesis kit (Roche Diagnostics, Mannheim, Germany). CTNS transcripts were amplified by PCR adding Taq DNA polymerase, PCR buffer with magnesium chloride (MgCl2), deoxyribonucleotide triphosphate (dNTP) (Invitrogen Life Technologies), and primers (Sigma-Aldrich, St. Louis, MO, USA) in accordance with the manufacturers’ instructions. Primers and PCR conditions for CTNS amplification are described in Table 2. PCR products were analyzed using a 1.5% agarose gel with GelRed™ staining (Biotium Inc., Hayward, CA, USA).
PCR fragments were extracted and purified with gel extraction kit (QIAGEN, Milan, Italy). Transcript sequencing was performed using primers detailed in Table 2 and dye terminator cycles sequencing (DTCS) Quick Start Mix (Beckman Coulter, Brea, CA, USA) at following conditions: 96°C for 30 s, 53°C for 30 s, and 60°C for 240 s for 37 cycles. After purification, samples were sequenced on a CEQ2000XL DNA Analysis System (Beckman Coulter).
Genomic DNA analysis
Genomic DNA was extracted from anticoagulated whole peripheral blood by QIAamp DNA blood kit (QIAGEN) in accordance with the manufacturer’s instructions. The CTNS gene was amplified using primers and PCR conditions detailed in Table 2. Direct sequencing was performed with the primers described in Table 2 and DTCS Quick Start Mix (Beckman Coulter) at conditions detailed above. Samples were sequenced on a CEQ2000XL DNA Analysis System (Beckman Coulter).
Results
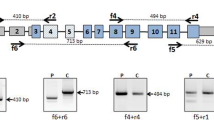
This study was performed on three patients with a clinical diagnosis of NC who had been previously screened for the CTNS gene mutations that revealed only one or no mutated alleles. Patients 1 and 2 are siblings. Previous sequence analysis had shown a heterozygous 57-kb deletion. No mutation was found on the other allele. To identify the second CTNS mutation, gene transcripts were analyzed from total RNA extracted from PBMCs. The PCR reaction yielded a 1,456-bp fragment spanning exons 2–12 that contained a 85-bp deletion corresponding to the entire exon 5 (Fig. 1). To highlight this deletion, CTNS transcripts were amplified using primers spanning exons 3–7 and were resolved by agarose gel electrophoresis, demonstrating a shorter band compared with control cDNA (Fig. 1). Skipping of exon 5 in these two patients causes a frameshift at position 141 bp, with a premature stop codon at position 160 bp. This splicing defect was further analyzed by sequencing the flanking intronic regions up to 300 bp upstream and downstream of exon 5. The analysis showed a point mutation at position –24 bp (c.141–24 T > C) of intron 4 that was not detected in 100 control chromosomes. The predicted protein generated by exon 5 skipping was truncated at position 53 after introducing six modified amino acids at the carboxy terminal end (wild-type cystinosin: 367 amino acids).

Detected DNA and messenger RNA (mRNA) changes. a Patients 1 and 2 had a T > C substitution at position –24, upstream of exon 5 (c.140–24 T > C) that was not detected by standard sequencing. This mutation causes a skipping of exon 5 in the CTNS gene transcripts, as demonstrated by direct sequencing and agarose gel electrophoresis after reverse transcriptase–polymerase chain reaction (RT-PCR) amplification of the region spanning exons 3–7. The smaller band (297 bp) corresponds to the amplicon lacking exon 5. b Patient 3 had a duplication of the genomic DNA sequence encompassing the last 73 bp of exon 9 and the first 193 bp of intron 9, shown in dark gray. This duplication causes an additional splicing of the mRNA transcript, with the incorporation of a portion of intron 9 followed by a repeat sequence corresponding to the duplicated portion of intron 9. By agarose gel electrophoresis using primers spanning exons 7 to 12, a larger 990-bp band is observed.
Genomic DNA analysis failed to reveal any CTNS gene mutations in patient 3. CTNS transcripts were amplified from skin fibroblasts and demonstrated a homozygous duplication located in the exon 9 region (Fig. 1). Specifically, the messenger RNA (mRNA) sequence of exon 9 was followed by a 75-bp sequence corresponding to a portion of intron 9, located 118 bp downstream to the exon donor splicing site and by a repeat sequence of the last 73 bp of exon 9. At the genomic level, this transcript corresponded to a duplication of the last 73 bp of exon 9 and the first 193 bp of intron 9, which introduces an additional splicing site that removes the first 118 bp of intron 9. When resolved on an agarose gel, the amplicon of patient 3 produced a larger band that included 148 additional nucleotides (Fig. 1). The predicted protein was truncated at position 232 by a frameshift that generated 5 mutated amino acids, followed by a stop codon.
Discussion
In this work, two novel mutations in the CTNS gene were detected by studying CTNS mRNA transcripts in patients in whom traditional genomic sequencing failed to detect mutations in one or both alleles. Specifically, a splicing defect and a DNA duplication were identified. Analysis of gene transcripts may be limited by tissue-specific expression patterns. Fortunately, the CTNS gene is ubiquitously expressed, and accumulation of increased cystine has been demonstrated in various tissues and cell lines, including fibroblasts, proximal tubular cells, and PBMCs [15–17].
In all our patients, the clinical diagnosis of NC was confirmed by measuring intraleucocyte cystine levels. The splicing defect in patients 1 and 2 was located in an intronic region that is not routinely investigated (T > C substitution 24 bp upstream of the splicing acceptor site of intron 4). This mutation is located at a hypothetical lariat branch site and probably plays an important role in the splicing of exon 5. Lariat branch sites are crucial for appropriate splicing of pre-mRNAs, and point mutations in these regions have been shown to cause abnormal mRNA splicing [18–20]. The 266-bp DNA duplication in patient 3 could not be detected using standard sequencing primers spanning exon 9 and its immediate neighboring intronic regions. In fact, this is the first DNA duplication described in patients with NC. Apart from point mutations and small insertion/deletions, all other mutations involve larger deletions [3, 6].
With this study, mutations in the CTNS gene were identified in all investigated Dutch patients, with the exception of one individual from whom RNA material was not available. Because the diagnosis of NC is substantiated by very specific findings, such as increased intraleucocyte cystine levels and corneal cystine crystals depositions, failure to discover mutations in these patients can be reasonably explained only by detection failures or by the existence of a second cystinotic gene. Our findings support the hypothesis that cystinosis is a monogenic disorder and suggest that CTNS transcripts should be studied when traditional genomic sequencing does not allow mutation detection. In addition, cystinosin-LKG is encoded by an alternative splicing of exon 12 and includes sequences that are located in the originally described 3′ untranslated region of the gene [5]. Although we have never observed mutations in this region (unpublished data), we also systematically include in our sequencing protocol the amplification of cystinosin-LKG-specific sequences.
References
Gahl WA, Thoene JG, Schneider JA (2002) Cystinosis. N Engl J Med 347:111–121
Gahl WA, Balog JZ, Kleta R (2007) Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med 147:242–250
Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, van’t Hoff W, Antignac C (1998) A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 18:319–324
Kalatzis V, Cherqui S, Antignac C, Gasnier B (2001) Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J 20:5940–5949
Taranta A, Petrini S, Palma A, Mannucci L, Wilmer MJ, De Luca V, Diomedi-Camassei F, Corallini S, Bellomo F, van den Heuvel LP, Levtchenko EN, Emma F (2008) Identification and subcellular localization of a new cystinosin isoform. Am J Physiol Renal Physiol 294:F1101–1108
Shotelersuk V, Larson D, Anikster Y, McDowell G, Lemons R, Bernardini I, Guo J, Thoene J, Gahl WA (1998) CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet 63:1352–1362
Heil SG, Levtchenko E, Monnens LA, Trijbels FJ, Van der Put NM, Blom HJ (2001) The molecular basis of Dutch infantile nephropathic cystinosis. Nephron 89:50–55
Kleta R, Anikster Y, Lucero C, Shotelersuk V, Huizing M, Bernardini I, Park M, Thoene J, Schneider J, Gahl WA (2001) CTNS mutations in African American patients with cystinosis. Mol Genet Metab 74:332–337
Kalatzis V, Cohen-Solal L, Cordier B, Frishberg Y, Kemper M, Nuutinen EM, Legrand E, Cochat P, Antignac C (2002) Identification of 14 novel CTNS mutations and characterization of seven splice site mutations associated with cystinosis. Hum Mutat 20:439–446
Kiehntopf M, Schickel J, Gönne B, Koch HG, Superti-Furga A, Steinmann B, Deufel T, Harms E (2002) Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis. Hum Mutat 20:237
Mason S, Pepe G, Dall’Amico R, Tartaglia S, Casciani S, Greco M, Bencivenga P, Murer L, Rizzoni G, Tenconi R, Clementi M (2003) Mutational spectrum of the CTNS gene in Italy. Eur J Hum Genet 11:503–508
Fernandez-Valero EM, Ballart A, Iturriaga C, Lluch M, Macias J, Vanier MT, Pineda M, Coll MJ (2005) Identification of 25 new mutations in 40 unrelated Spanish Niemann-Pick type C patients: genotype-phenotype correlations. Clin Genet 68:245–254
Alcántara-Ortigoza MA, Belmont-Martínez L, Vela-Amieva M, González-Del Angel A (2008) Analysis of the CTNS gene in nephropathic cystinosis Mexican patients: report of four novel mutations and identification of a false positive 57-kb deletion genotype with LDM-2/exon 4 multiplex PCR assay. Genet Test 12:409–414
Levtchenko EN, Wilmer MJ, Janssen AJ, Koenderink JB, Visch HJ, Willems PH, de Graaf-Hess A, Blom HJ, van den Heuvel LP, Monnens LA (2006) Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res 59:287–292
Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD (1982) Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217:1263–1265
Jonas AJ, Greene AA, Smith ML, Schneider JA (1982) Cystine accumulation and loss in normal, heterozygous, and cystinotic fibroblasts. Proc Natl Acad Sci USA 79:4442–4445
Wilmer MJ, de Graaf-Hess A, Blom HJ, Dijkman HB, Monnens LA, van den Heuvel LP, Levtchenko EN (2005) Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem Biophys Res Commun 337:610–614
Kuivenhoven JA, Weibusch H, Pritchard PH, Funke H, Benne R, Assmann G, Kastelein JJ (1996) An intronic mutation in a lariat branchpoint sequence is a direct cause of an inherited human disorder (fish-eye disease). J Clin Invest 98:358–364
Baralle D, Baralle M (2005) Splicing in action: assessing disease causing sequence changes. J Med Genet 42:737–748
Di Leo E, Panico F, Tarugi P, Battisti C, Federico A, Calandra S (2004) A point mutation in the lariat branch point of intron 6 of NPC1 as the cause of abnormal pre-mRNA splicing in Niemann-Pick type C disease. Hum Mutat 24:440
Acknowledgments
This work was supported by a grant from the Cystinosis Research Foundation (Irvine, CA, USA) and the Cystinosis Research Network.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Taranta, A., Wilmer, M.J., van den Heuvel, L.P. et al. Analysis of CTNS gene transcripts in nephropathic cystinosis. Pediatr Nephrol 25, 1263–1267 (2010). https://doi.org/10.1007/s00467-010-1502-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-010-1502-5