
Abstract
The severity of renal involvement is the major factor determining the long-term outcome of children with Henoch-Schönlein purpura (HSP) nephritis (HSPN). Approximately 40% children with HSP develop nephritis, usually within 4 to 6 weeks after the initial onset of the typical purpuric rashes. Although the pathogenetic mechanisms are still not fully delineated, several studies suggest that galactose-deficient IgA1 (Gd-IgA1) is recognized by anti-glycan antibodies, leading to the formation of the circulating immune complexes and their mesangial deposition that induce renal injury in HSPN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Henoch-Schönlein purpura (HSP) is predominantly a childhood vasculitic disease, characterized by the presence of immunoglobulin A1 (IgA1) dominant immune deposits in the small vessels [1, 2]. While HSP is often a self-limited condition, approximately 40% pediatric patients develop nephritis within 4 to 6 weeks of the initial presentation [2]. Some children with HSP nephritis (HSPN) subsequently have an episode or recurrent episodes of macroscopic hematuria, frequently associated with upper respiratory tract infection without the other clinical features of HSP [3, 4]. Thus, these children’s clinical phenotype changes to one of IgA nephropathy (IgAN). As the renal histologic and immunofluorescence microscopy findings in HSPN are indistinguishable from those seen in patients with IgAN [5], it has long been speculated that HSPN and IgAN share common pathogenetic mechanisms and may represent different ends of a continuous spectrum of disease [3]. Knowledge acquired from studies of patients with IgAN may also provide important insights into the pathogenesis of HSPN.
The role of IgA in pathogenesis of HSPN
The postulation that HSPN is a systemic immune-complex mediated disease is supported by the clinical or histological recurrences of HSPN in some patients after transplantation [6, 7]. Although detailed pathogenic mechanisms of HSPN have not been fully elucidated, perturbations in the immune system, including elevations in serum levels of IgA1, IgA1-containing circulating immune complexes and IgA-rheumatoid factors have been documented for patients with HSP [8–11]. Elevated serum levels of IgA and IgA-containing immune complexes were observed in patients with HSPN [12]. Furthermore, it was noted that all HSP patients have IgA1-circulating immune complexes of small molecular mass, but only those with nephritis have additional large-molecular-mass IgA1-IgG-containing circulating immune complexes [13].
The risk for progression of HSPN was associated with increasing mean proteinuria levels during follow-up and was greater in adults and females [14]. IgA1-containing immune complexes are excreted in elevated amounts in the urine in patients with IgAN and HSPN and may provide a specific marker for disease activity and/or severity in these patients [15, 16].
Levels of serum IgA may not be a diagnostic marker of HSPN
Although serum IgA levels are higher in children with HSP/HSPN than in controls [17], this serum abnormality does not constitute a sensitive diagnostic marker of HSP or HSPN. In a recent retrospective study from Brazil, over 40% children with HSP had elevated serum IgA levels at presentation [18], but the difference in serum IgA levels between patients with and without nephritis was not statistically significant. On the other hand, in spite of high levels of circulating serum IgA in patients with multiple myeloma, HSPN cases have rarely been reported in these patients [19]. These observations argue against the assumption that high serum IgA alone predisposes patients to nephritis.
Abnormal glycosylation of IgA1 and its role in the pathogenesis of HSPN
HSPN is similar to IgAN in that IgA1, but not IgA2, is found in the circulating immune complexes and in mesangial immune deposits [12, 20, 21]. Only humans and higher primates have IgA1 [22]. As depicted in Fig. 1, the human IgA1 molecule has a hinge region containing up to six O-linked glycan chains [23, 24] consisting of N-acetylgalactosamine (GalNAc), usually with a β1,3-linked galactose (Gal) attached to it [25, 26]. GalNAc and/or Gal may or may not be sialylated and serum IgA1 from normal healthy subjects predominantly has the Gal-GalNAc disaccharide in its mono- and di-sialylated forms [27, 28].

IgA1 and its hinge region with O-linked glycans (white circles) and N-linked glycans (black circles). Underlined amino acids denote usual sites of attachment of as many as five O-linked glycans [99]. Examples of galactose-deficient (in red color) and galactosylated (in blue color) O-linked glycans in the hinge region of human circulatory IgA1 are shown at the bottom. GalNAc, N-acetylgalactosamine; Gal, galactose; SA, sialic acid
Similarly as in patients with IgAN, increased galactose-deficient IgA1 (Gd-IgA1) levels have been detected in patients with HSPN [29, 30], but not in subjects with other glomerular diseases. With several lines of evidence, Gd-IgA1 now assumes a pivotal role in the pathogenesis of both IgAN and HSPN. Using GalNAc-specific lectin from Vicia villosa, patients with HSPN were shown to have increased lectin binding, indicating elevated levels of Gd-IgA1, while the levels of Gd-IgA1 in patients with HSP without nephritis were similar to healthy controls [29]. We recently reported, using GalNAc-specific lectin from Helix aspersa, that the serum levels of Gd-IgA1 were higher in children with IgAN and HSPN compared to healthy controls and patients with C1q nephropathy [30]. Importantly, the median levels of serum Gd-IgA1 in children with HSP without nephritis did not significantly differ from those in healthy controls [30]. These data corroborate a potential pathogenic role for Gd-IgA1 in HSPN. Moreover, IgA molecules in the skin immune deposits of patients with HSP regardless of renal involvement are exclusively of IgA1 subclass [31], but there have been no studies on the glycosylation of these IgA1 molecules.
The biosynthetic origin of Gd-IgA1
There are two major sites with IgA1-producing cells: mucosal tissues and bone marrow. Both have been suggested as possible sites of production of the aberrantly glycosylated IgA1 [32–39]. Reduced galactosylation of IgA1 O-glycans has been suggested by the results of lectin ELISA [29]; these finding correlated with the observation of reduced activity of β1,3-galactosyltransferase in the peripheral B cells of patients with IgAN and HSPN [40–42]. The primary defect that leads to the production of such abnormally glycosylated IgA1 is probably heritable [43]. Moreover, Suzuki et al. have recently immortalized and cloned B cells from patients with IgAN that synthesized and secreted an undergalactosylated form of IgA1 [44]. Such glycosylation defects, due to complex changes in expression of specific glycosyltransferases with reduced expression of β1,3-galactosyltransferase and elevated expression of GalNAc-specific α2,6-sialyltransferase, were detected in patients with IgAN and HSPN, but not in patients with HSP without nephritis or healthy controls [40, 44, 45]. These data suggest that premature sialylation may contribute to the aberrant IgA1 O-glycosylation in IgAN and these cell lines provide a new model for future studies on the mechanisms leading to aberrant IgA1 O-glycosylation in IgAN and HSPN.
Interestingly, IgG in the serum and IgG produced by cloned IgG-secreting cells from patients with HSPN had elevated levels of IgG against Gd-IgA1 compared to those from HSP patients without nephritis and controls [40, 46]. The possible origin and role of these anti-glycan antibodies in the pathogenesis of HSPN needs further study. However, they now appear to be the major factor responsible for the formation of the nephritogenic immune complexes, consistent with the previously described large-molecular mass IgA-IgG complexes in the circulation of patients with HSPN [13].
Which sites on IgA1 contain aberrantly glycosylated O-glycans?
The assays based on the binding of GalNAc-specific lectins to IgA1 indicated the presence of aberrant glycosylation in the IgA1 molecules; the assay itself cannot assess whether all the sites contain these anomalies or whether only specific sites are affected. Analysis of the IgA1 molecules from patients with IgAN and HSPN shed some light on the localization of aberrant glycans. Use of bacterial IgA-specific proteases together with lectin western blotting and mass spectrometry suggested presence of Gal-deficient O-glycans (terminal GalNAc or sialylated GalNAc - see Fig. 1) at T228/S230 and S232 in the hinge region of IgA1 in patients with IgAN; immune complexes containing such IgA1 bound with IgG specific to Gal-deficient IgA1 were able to stimulate proliferation of cultured human mesangial cells [20, 40, 47–49].
Sialylation of IgA1 molecules in IgAN and HSPN
As sialic acid carries a high negative charge, the degree of sialylation may alter interactions of IgA1 with other molecules and thus affect clearance of IgA1 and IgA1-containing immune complexes. Although removal of sialic acid has been shown to increase the aggregation of IgA1 molecules [50, 51], in a recent study by Leung et al., neuraminidase treatment (removal of sialic acid) significantly reduced the binding capability of polymeric IgA1 from IgAN patients to human mesangial cells in vitro [52]. It is therefore of interest that a portion of IgA1 from patients with IgAN is Gal-deficient and over sialylated [44, 53].
Possible mechanisms involving Gd-IgA1 in the development of HSPN
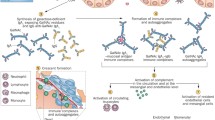
Figure 2 depicts a proposed mechanism of how aberrantly glycosylated IgA1 may become nephritogenic. Polymeric Gd-IgA1 molecules are recognized by naturally occurring anti-glycan IgA1 or IgG and circulating immune complexes are formed [20, 40, 50, 54–57]. Due to their size, Gd-IgA1-containing immune complexes are less efficiently taken up by the asialoglycoprotein receptor in the liver and catabolized and their amounts increase in the circulation [58, 59]. These complexes may then deposit in the renal mesangium and incite glomerular injury, likely due to the binding to mesangial cells leading to cellular activation. Consequently, mesangial cells start to proliferate and overproduce extracellular matrix components, cytokines and chemokines [29, 60–62]. CD71 (transferrin receptor), was found to be a receptor for polymeric IgA1 on the mesangial cells, and could provide a mechanism for how the Gd-IgA immune complexes bind to the mesangial cells [63]. Expression of CD71 on the mesangial cell surface was increased in pediatric patients with IgAN and HSPN as compared to children with other forms of glomerular disease [63]. The intensity of CD71 expression associated with the degree of cellular proliferation rather then the intensity of the IgA staining. Hence, the processes involving IgA1 mesangial deposition, the molecular mechanisms underlying the interaction of IgA1-containing circulating immune complexes and cell receptors that lead to mesangial cell activation and initiation of glomerular inflammatory processes need to be more completely delineated.

Possible role of IgA1 and IgA1-containing immune complexes in the pathogenesis of HSPN
Composition of immune deposits in HSPN
While Gd-IgA1-containing immune complexes are increasingly recognized as a major player in the pathogenesis of HSPN [29, 30], other components in these immune complexes have also been examined for their possible pathogenetic roles. Although it has been reported that IgG contents in the IgA-containing circulating immune complexes from patients with HSPN were higher than those with IgAN [64, 65], IgG deposition is not a universal finding in renal biopsies from patients with either disease [13, 66–68]. Likewise, IgM deposits were inconsistently demonstrated in the glomerular deposits of patients with HSPN [67, 68]. Thus, the roles of IgG and IgM in pathogenesis of HSPN still need further elucidation. J-chain has also been demonstrated on renal biopsies from patients with IgAN, consistent with the presence of polymeric IgA1 [67, 68].
Glomerular depositions of other components, including kappa and lambda light chains, are also variably demonstrated in HSPN. In patients with IgAN, lambda light chains were found predominantly over kappa light chains [69, 70]. However, the ratio of lambda chains to kappa chains in the mesangial deposits remains about equal in patients with HSPN [71]. The discrepancy in synthesis of different types of light chains in patients with IgAN and HSPN, and their implications in pathogenesis, is not clear.
Potential role of complement system in the pathogenesis of HSPN
Complement activation appears to play an important role in the pathogenesis of IgAN and HSPN, as glomerular complement activation may initiate the inflammatory cascade and enhance glomerular injury [72]. Although hypocomplementemia has been reported in some patients with HSPN [73, 74], it is usually transient and not related to the severity of the diseases. Since the first report on higher incidence of either C4A or C4B null variants, or both, in patients with IgAN and HSP [75], C4A and C4B deficiencies had also been described in patients with HSPN [76, 77]. In Iceland, the frequency of C4B null alleles was significantly increased in children with HSP as compared to controls [78]. The role of partial or complete C4 isotype deficiencies in pathogenesis of HSPN may relate to impairment in the ability to solubulize and/or clear immune complexes [79], but the precise mechanisms involved remain speculative.
Although it has long been known that the alternative complement pathway is activated in patients with IgAN and HSPN [80], attention has shifted to the lectin pathway. This third pathway of complement activation is initiated by mannose-binding lectin (MBL). MBL also forms complexes with MBL-associated serine protease-1 (MASP-1), MASP-2 and MASP-3 [81–84]. Recent studies suggested that complement activation occurs through both the alternate and lectin pathways in patients with IgAN [81, 82].
The long pentraxin 3 (PTX3), a complement related protein, has been detected in the renal tissue of patients with glomerulonephritis [85]. In renal biopsies from patients with IgAN, intense staining for PTX3 was observed in the expanded mesangial areas and was localized to the glomerular mesangial and endothelial cells. Normal renal tissue and biopsies from patients with other glomerular nephropathies were negative for PTX3 expression in glomeruli. Cultured human mesangial cells synthesized PTX3 when stimulated with TNF-α and IgA, and exhibited specific binding for recombinant PTX3 [85]. These observations suggest potential role for PTX3 in the modulation of glomerular injury in IgAN and likely also in HSPN. PTX3 appears to be involved in the classical and not the alternative pathway of complement activation [86]. Thus, its role in pathogenesis of HSPN, where the complement activation is alternative and/or lectin pathway mediated [81, 82], will require further clarification.
Possible role of cytokines in the pathogenesis of HSPN
In cell lines isolated from patients with IgAN, but not in those from controls, cytokine stimulation reduced the expression of β1,3-galactosyltransferase and its molecular chaperone Cosmc, but increased the expression of α2,6- GalNAc-sialytransferase II. As a result, the synthesis of sialylated GalNAc was enhanced [87], pointing to a potential role of some cytokines in the pathogenesis of IgAN; these aspects in HSPN require further investigations.
Other possible pathogenic mechanisms of HSPN
Masuda et al. showed that nephritis-associated plasmin receptor (NAPlr), a group A streptococcal antigen, may also have pathogenetic role in a subset of patients with HSPN [88]. Among 33 children with biopsy proven HSPN, 30% had segmental or global mesangial deposition of NAPlr antigen, comparing to 3% in other children with non-HSPN glomerular diseases (half of these children had IgAN). The exact pathophysiologic mechanism, if any, and the relationship between NAPlr and HSPN, needs further investigation.
In a study by Davin et al, 22 children with HSPN were compared to 16 children with IgAN. In their cohort, elevated plasma IgE levels were more commonly found in patients with HSPN (77% versus 44%) [89]. They hypothesised that the IgA containing immune complexes could enhance local IgE production via stimulation the dermal and intestinal mast cells. Deposition of the IgA immune complexes was further enhanced with the subsequent increase in local capillary permeability [89]. Notwithstanding the higher incidence of elevated plasma level of IgE in patients with HSPN, the pathogenetic roles of IgE remains unclear, as the mast cell is not usually found in the mesangium [90, 91].
Eosinophil activation has also been proposed to play a role in the pathogenesis of HSPN [92–94]. Children with HSP, compared to those with IgAN or healthy controls, have higher levels of serum eosinophil cationic protein (ECP) [93]. And the levels were even higher in patients with HSPN. In a recent study from Japan, patients with HSPN were also demonstrated to have higher serum concentrations of ECP and interleukin-5 [93]. In another study from China, serum levels of ECP were higher only when the patients had active HSPN [94]. These studies suggested that ECP might have a role in the initiation of nephritis in patients with HSP.
Serum IgA anti-cardiolipin and anti-phosphatidylserine-prothrombin complexes were both elevated and associated with the severity of proteinuria in a group of Japanese adult patients with HSP [95]. The role of these IgA antibodies in the pathogenesis in HSP and HSPN is not clear.
Recently, renal expression of alpha-smooth muscle actin (α-SMA) has also been associated with progression of renal injury in patients with HSPN [96]. Thirty five patients in Japan with biopsy proven HSPN were studied in 3 groups: 1) nephritis with histological class (classification system used by the International Study of Kidney Disease in Children [97]) of stage II or less, 2) histological stage III or greater and a good prognosis, and 3) histological stage II or greater and poor prognosis. All patients, except those in group 1, had repeated biopsy during the course of follow-up. The authors found that the mean scores for glomerular and interstitial α-SMA staining at first biopsy were higher in HSPN patients with crescents. And at second biopsy, α-SMA expressions were also higher in patients with poor prognosis. Although α-SMA is the predominant actin isoform within vascular smooth muscle and the mechanism of phenotypic changes of mesangial cells and increased expression of α-SMA in patients with HSPN is unclear, these observations did suggest that increased renal α-SMA expression may be an early histological indicator of progression of HSPN. Similarly, increased expression of α-SMA in the tubulointerstitial area, but not in glomeruli, was associated with poor prognosis of patients with IgAN [98].
Conclusions
HSPN is a common pediatric renal disease with potential long-term morbidity. Co-operative efforts of clinical and basic research scientists have provided valuable information and insights on the pathogenetic mechanisms of HSPN. Although these mechanisms are far from being completely understood, multiple possible players have been identified. Recent data suggest that Gd-IgA1 is likely to play a pivotal role in the formation of nephritogenic immune complexes (Fig. 2) and further investigations as to its role are ongoing.
References
Niaudet P, Habib R (1994) Schönlein-Henoch purpura nephritis: pronostic factors and therapy. Ann Med Interne (Paris) 145:577–580
Saulsbury FT (2007) Clinical update: Henoch-Schönlein purpura. Lancet 369:976–978
Waldo FB (1988) Is Henoch-Schönlein purpura the systemic form of IgA nephropathy? Am J Kidney Dis 12:373–377
Nakamoto Y, Asano Y, Dohi K, Fujioka M, Iida H, Kida H, Kibe Y, Hattori N, Takeuchi J (1978) Primary IgA glomerulonephritis and Schönlein-Henoch purpura nephritis: Clinicopathological and immunohistological characteristics. Q J Med 47:495–516
Evans DJ, Williams DG, Peters DK, Sissons JG, Boulton-Jones JM, Ogg CS, Cameron JS, Hoffbrand BI (1973) Glomerular deposition of properdin in Henoch-Schönlein syndrome and idiopathic focal nephritis. Br Med J 3:326–328
Meulders Q, Pirson Y, Cosyns JP, Squifflet JP, van Ypersele de Strihou C (1994) Course of Henoch-Schönlein nephritis after renal transplantation. Report on ten patients and review of the literature. Transplantation 58:1179–1186
Soler MJ, Mir M, Rodriguez E, Orfila A, Munne A, Vázquez S, Lloveras J, Puig JM (2005) Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 37:3705–3709
Saulsbury FT (1986) IgA rheumatoid factor in Henoch-Schönlein purpura. J Pediatr 108:71–76
Knight JF (1990) The rheumatic poison: a survey of some published investigations of the immunopathogenesis of Henoch-Schönlein purpura. Pediatr Nephrol 4:533–541
Fervenza FC (2003) Henoch-Schönlein purpura nephritis. Int J Dermatol 42:170–177
Davin JC, Ten Berge IJ, Weening JJ (2001) What is the difference between IgA nephropathy and Henoch-Schönlein purpura nephritis? Kidney Int 59:823–834
Coppo R, Basolo B, Piccoli G, Mazzucco G, Bulzomì MR, Roccatello D, De Marchi M, Carbonara AO, Barbiano di Belgiojoso G (1984) IgA1 and IgA2 immune complexes in primary IgA nephropathy and Henoch-Schönlein nephritis. Clin Exp Immunol 57:583–590
Levinsky RJ, Barratt TM (1979) IgA immune complexes in Henoch-Schönlein purpura. Lancet 2:1100–1103
Coppo R, Andrulli S, Amore A, Gianoglio B, Conti G, Peruzzi L, Locatelli F, Cagnoli L (2006) Predictors of outcome in Henoch-Schönlein nephritis in children and adults. Am J Kidney Dis 47:993–1003
Suzuki H, Suzuki Y, Lyas CN, Kirksey C, Hall S, Moldoveanu Z, Wyatt RJ, Mestecky J, Tomino Y, Novak J, Julian A (2008) Urinary polypeptide biomarkers of IgA nephropathy. J Am Soc Nephrol 19:665A
Julian BA, Wittke S, Novak J, Good DM, Coon JJ, Kellmann M, Zürbig P, Schiffer E, Haubitz M, Moldoveanu Z, Calcatera SM, Wyatt RJ, Sýkora J, Sládková E, Hes O, Mischak H, McGuire BM (2007) Electrophoretic methods for analysis of urinary polypeptides in IgA-associated renal diseases. Electrophoresis 28:4469–4483
Lin SC, Tsai MJ, Huang MT, Wu KH, Wang LH, Chiang BL (1998) Immunological studies of children with anaphylactoid purpura. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 39:247–252
de Almeida JL, Campos LM, Paim LB, Leone C, Koch VH, Silva CA (2007) Renal involvement in Henoch-Schönlein purpura: a multivariate analysis of initial prognostic factors. J Pediatr (Rio J) 83:259–266
Zickerman AM, Allen AC, Talwar V, Olczak SA, Brownlee A, Holland M, Furness PN, Brunskill NJ, Feehally J (2000) IgA myeloma presenting as Henoch-Schönlein purpura with nephritis. Am J Kidney Dis 36:E19
Novak J, Moldoveanu Z, Renfrow MB, Yanagihara T, Suzuki H, Raska M, Hall S, Brown R, Huang WQ, Goepfert A, Kilian M, Poulsen K, Tomana M, Wyatt RJ, Julian BA, Mestecky J (2007) IgA nephropathy and Henoch-Schoenlein purpura nephritis: aberrant glycosylation of IgA1, formation of IgA1-containing immune complexes, and activation of mesangial cells. Contrib Nephrol 157:134–138
Conley ME, Cooper MD, Michael AF (1980) Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J Clin Invest 66:1432–1436
Mestecky J, Moro I, Kerr MA, Woof JM (2005) Mucosal immunoglobulins. In: Mestecky J, Bienenstock J, Lamm ME, Mayer L, McGhee JR, Strober W (eds) Mucosal immunology, 3rd edn. Elsevier Academic Press, Amsterdam, pp 153–181
Tarelli E, Smith AC, Hendry BM, Challacombe SJ, Pouria S (2004) Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr Res 339:2329–2335
Renfrow MB, MacKay CL, Chalmers MJ, Julian BA, Mestecky J, Kilian M, Poulsen K, Emmett MR, Marshall AG, Novak J (2007) Analysis of O-glycan heterogeneity in IgA1 myeloma proteins by Fourier transform ion cyclotron resonance mass spectrometry: Implications for IgA nephropathy. Anal Bioanal Chem 389:1397–1407
Baenziger J, Kornfeld S (1974) Structure of the carbohydrate units of IgA1 immunoglobulin II. Structure of the O-glycosidically linked oligosaccharide units. J Biol Chem 249:7270–7281
Field MC, Dwek RA, Edge CJ, Rademacher TW (1989) O-linked oligosaccharides from human serum immunoglobulin A1. Biochem Soc Trans 17:1034–1035
Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, Rudd PM, Woof JM, Dwek RA (1998) The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J Biol Chem 273:2260–2272
Novak J, Tomana M, Kilian M, Coward L, Kulhavy R, Barnes S, Mestecky J (2000) Heterogeneity of O-glycosylation in the hinge region of human IgA1. Mol Immunol 37:1047–1056
Allen AC, Willis FR, Beattie TJ, Feehally J (1998) Abnormal IgA glycosylation in Henoch-Schönlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant 13:930–934
Lau KK, Wyatt RJ, Moldoveanu Z, Tomana M, Julian BA, Hogg RJ, Lee JY, Huang WQ, Mestecky J, Novak J (2007) Serum levels of galactose-deficient IgA in children with IgA nephropathy and Henoch-Schönlein purpura. Pediatr Nephrol 22:2067–2072
Egan CA, Taylor TB, Meyer LJ, Petersen MJ, Zone JJ (1999) IgA1 is the major IgA subclass in cutaneous blood vessels in Henoch-Schönlein purpura. Br J Dermatol 141:859–862
van den Wall Bake AW, Daha MR, Radl J, Haaijman JJ, Van der Ark A, Valentijn RM, Van Es LA (1988) The bone marrow as production site of the IgA deposited in the kidneys of patients with IgA nephropathy. Clin Exp Immunol 72:321–325
van den Wall Bake AW, Daha MR, Evers-Schouten J, van Es LA (1988) Serum IgA and the production of IgA by peripheral blood and bone marrow lymphocytes in patients with primary IgA nephropathy: evidence for the bone marrow as the source of mesangial IgA. Am J Kidney Dis 12:410–414
Feehally J, Allen AC (1999) Structural features of IgA molecules which contribute to IgA nephropathy. J Nephrol 12:59–65
Buck KS, Smith AC, Molyneux K, El-Barbary H, Feehally J, Barratt J (2008) B-cell O-galactosyltransferase activity, and expression of O-glycosylation genes in bone marrow in IgA nephropathy. Kidney Int 73:1128–1136
Smith AC, Molyneux K, Feehally J, Barratt J (2006) O-glycosylation of serum IgA1 antibodies against mucosal and systemic antigens in IgA nephropathy. J Am Soc Nephrol 17:3520–3528
Béné MC, Faure GC, Hurault de Ligny B, de March AK (2004) Clinical involvement of the tonsillar immune system in IgA nephropathy. Acta Otolaryngol Suppl 555:10–14
Béné MC, Kennel A, Renoult E, Kessler M, Faure GC (1995) Altered mucosal immunity in IgA nephropathy investigated using the ELISA spot method in peripheral blood. Contrib Nephrol 111:123–128
Béné MC, Faure GC (1988) Mesangial IgA in IgA nephropathy arises from the mucosa. Am J Kidney Dis 12:406–409
Suzuki H, Moldoveanu Z, Wyatt R, Stacy Hall1, Brown R, Julian B, Tomino Y, Mestecky J, Novak J (2008) Aberrantly glycosylated IgA1 and anti-glycan IgG antibodies are elevated in patients with active Henoch-Schoenlein Purpura nephritis. J Am Soc Nephrol 19:658A
Allen AC, Topham PS, Harper SJ, Feehally J (1997) Leucocyte β 1, 3 galactosyltransferase activity in IgA nephropathy. Nephrol Dial Transplant 12:701–706
Barratt J, Feehally J (2005) IgA nephropathy. J Am Soc Nephrol 16:2088–2097
Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, Mestecky J, Novak J, Julian BA (2008) Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol 19:1008–1014
Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, Julian BA, Tomana M, Wyatt RJ, Edberg JC, Alarcón GS, Kimberly RP, Tomino Y, Mestecky J, Novak J (2008) IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest 118:629–639
Raska M, Moldoveanu Z, Suzuki H, Brown R, Kulhavy R, Andrasi J, Hall S, Vu HL, Carlsson F, Lindahl G, Tomana M, Julian BA, Wyatt RJ, Mestecky J, Novak J (2007) Identification and characterization of CMP-NeuAc:GalNAc-IgA1 α2, 6-sialyltransferase in IgA1-producing cells. J Mol Biol 369:69–78
Suzuki H, Moldoveanu Z, Hall S, Brown R, Julian BA, Wyatt RJ, Lee JY, Tomana M, Tomino Y, Mestecky J, Novak J (2007) Immunoglobulin-producing cell lines from patients with Henoch-Schönlein purpura nephritis secrete aberrantly-glycosylated IgA1 and anti-glycan IgG antibodies. J Am Soc Nephrol 18:188A
Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, Julian BA, Wyatt RJ, Mestecky J (2005) IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int 67:504–513
Moore JS, Kulhavy R, Tomana M, Moldoveanu Z, Suzuki H, Brown R, Hall S, Kilian M, Poulsen K, Mestecky J, Julian BA, Novak J (2007) Reactivities of N-acetylgalactosamine-specific lectins with human IgA1 proteins. Mol Immunol 44:2598–2604
Suzuki H, Hall S, Renfrow MB, Moldoveanu Z, Lyas CN, Julian BA, Kilian M, Poulsen K, Mestecky J, Novak J (2008) Bacterial IgA proteases and analysis of aberrant O-glycosylation of IgA1 in patients with IgA nephropathy. J Am Soc Nephrol 19:658A
Kokubo T, Hiki Y, Iwase H, Tanaka A, Toma K, Hotta K, Kobayashi Y (1998) Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol 9:2048–2054
Iwase H, Tanaka A, Hiki Y, Kokubo T, Sano T, Ishii-Karakasa I, Toma K, Kobayashi Y, Hotta K (1999) Mutual separation of hinge-glycopeptide isomers bearing five N-acetylgalactosamine residues from normal human serum immunoglobulin A1 by capillary electrophoresis. J Chromatogr B Biomed Sci Appl 728:175–183
Leung JC, Chan LY, Tang SC, Tam PC, Fenn J, Lai KN (2007) Glycosylation profile of differently charged IgA1 and their binding characteristics to cultured mesangial cells in IgA nephropathy. Nephron Exp Nephrol 107:e107–e118
Leung JC, Tang SC, Chan DT, Lui SL, Lai KN (2002) Increased sialylation of polymeric λ-IgA1 in patients with IgA nephropathy. J Clin Lab Anal 16:11–19
Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J (1999) Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest 104:73–81
Mestecky J, Suzuki H, Yanagihara T, Moldoveanu Z, Tomana M, Matousovic K, Julian BA, Novak J (2007) IgA Nephropathy: Current Views of Immune Complex Formation. In: IgA Nephropathy Today (Tomino Y, ed) Contributions to Nephrology Vol. 157, Karger Corporation, Basel, pp 56–63
Suzuki H, Moldoveanu Z, Hall S, Brown R, Julian BA, Wyatt RJ, Tomana M, Tomino Y, Novak J, Mestecky J (2007) IgA Nephropathy: Characterization of IgG Antibodies Specific for Galactose-deficient IgA1. In: IgA Nephropathy Today (Tomino Y, ed) Contributions to Nephrology Vol. 157, Karger Corporation, Basel, pp 129–133
Novak J, Julian BA, Tomana M, Mestecky J (2008) IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol 28:78–87
Mestecky J, Tomana M, Crowley-Nowick PA, Moldoveanu Z, Julian BA, Jackson S (1993) Defective galactosylation and clearance of IgA1 molecules as a possible etiopathogenic factor in IgA nephropathy. Contrib Nephrol 104:172–182
Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, Vrtovsnik F, Haddad E, Chintalacharuvu KR, Monteiro RC (2004) Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol 15:622–634
Davin JC, Weening JJ (2003) Diagnosis of Henoch-Schönlein purpura: renal or skin biopsy? Pediatr Nephrol 18:1201–1203
Novak J, Vu HL, Novak L, Julian BA, Mestecky J, Tomana M (2002) Interactions of human mesangial cells with IgA and IgA-containing immune complexes. Kidney Int 62:465–475
Kokubo T, Hiki Y, Iwase H, Horii A, Tanaka A, Nishikido J, Hotta K, Kobayashi Y (1997) Evidence for involvement of IgA1 hinge glycopeptide in the IgA1-IgA1 interaction in IgA nephropathy. J Am Soc Nephrol 8:915–919
Haddad E, Moura IC, Arcos-Fajardo M, Macher MA, Baudouin V, Alberti C, Loirat C, Monteiro RC, Peuchmaur M (2003) Enhanced expression of the CD71 mesangial IgA1 receptor in Berger disease and Henoch-Schönlein nephritis: association between CD71 expression and IgA deposits. J Am Soc Nephrol 14:327–337
Kauffmann RH, Van Es LA, Daha MR (1981) The specific detection of IgA in immune complexes. J Immunol Methods 40:117–129
Cederholm B, Linne T, Wieslander J, Bygren P, Heinegård D (1991) Fibronectin-immunoglobulin complexes in the early course of IgA and Henoch-Schönlein nephritis. Pediatr Nephrol 5:200–204
Conley ME, Cooper MD, Michael AF (1980) Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J Clin Invest 66:1432–1436
Tomino Y, Endoh M, Suga T, Miura M, Kaneshige H, Nomoto Y, Sakai H (1982) Prevalence of IgAl deposits in Henoch-Schoenlein purpura (HSP) nephritis. Tokai J Exp Clin Med 7:527–532
Lomax-Smith JD, Zabrowarny LA, Howarth GS, Seymour AE, Woodroffe AJ (1983) The immunochemical characterization of mesangial IgA deposits. Am J Pathol 113:359–364
Lai KN, Chui SH, Lai FM, Lam CW (1988) Predominant synthesis of IgA with lambda light chain in IgA nephropathy. Kidney Int 33:584–589
Lai KN, To WY, Li PK, Leung JC (1996) Increased binding of polymeric lambda-IgA to cultured human mesangial cells in IgA nephropathy. Kidney Int 49:839–845
Emancipator SN (1993) Primary and secondary forms of IgA nephritis and Schönlein-Henoch syndrome,. In: Pathology of the Kidney; edited by Heptinstall RH Toronto, London, Little, Brown pp 389–476
Wyatt RJ, Kanayama Y, Julian BA, Negoro N, Sugimoto S, Hudson EC, Curd JG (1987) Complement activation in IgA nephropathy. Kidney Int 31:1019–1023
Motoyama O, Iitaka K (2005) Henoch-Schönlein purpura with hypocomplementemia in children. Pediatr Int 47:39–42
Garcia-Fuentes M, Martin A, Chantler C, Williams DG (1978) Serum complement components in Henoch-Schönlein purpura. Arch Dis Child 53:417–419
McLean RH, Wyatt RJ, Julian BA (1984) Complement phenotypes in glomerulonephritis: increased frequency of homozygous null C4 phenotypes in IgA nephropathy and Henoch-Schönlein purpura. Kidney Int 26:855–860
Wyatt RJ, Julian BA, Woodford SY, Wang C, Roberts J, Thompson JS, Christenson MJ, McLean RH (1991) C4A deficiency and poor prognosis in patients with IgA nephropathy. Clin Nephrol 36:1–5
Ault BH, Stapleton FB, Rivas ML, Waldo FB, Roy S 3rd, McLean RH, Bin JA, Wyatt RJ (1990) Association of Henoch-Schönlein purpura glomerulonephritis with C4B deficiency. J Pediatr 117:753–755
Stefansson Thors V, Kolka R, Sigurdardottir SL, Edvardsson VO, Arason G, Haraldsson A (2005) Increased Frequency of C4B*Q0 Alleles in Patients with Henoch-Schönlein Purpura. Scand J Immunol 61:274–278
Atkinson JP (1989) Complement deficiency: predisposing factor to autoimmune syndromes. Clin Exp Rheumatol 7(Suppl 3):S95–S101
Wyatt RJ (1993) The complement system in IgA nephropathy and Henoch-Schönlein purpura: functional and genetic aspects. Contrib Nephrol 104:82–91
Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M, Fujita T (1998) Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrol Dial Transplant 13:1984–1990
Matsuda M, Shikata K, Wada J, Sugimoto H, Shikata Y, Kawasaki T, Makino H (1998) Deposition of mannan binding protein and mannan binding protein-mediated complement activation in the glomeruli of patients with IgA nephropathy. Nephron 80:408–313
Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR (2006) Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol 17:1724–1734
Dahl MR, Thiel S, Willis AC, Vorup-Jensen T, Christensen T, Petersen SV, Jensenius JC (2000) Mannan-binding lectin associated serine protease 3 (MASP-3)-a new component of the lectin pathway of complement activation. Immunopharmacology 49:79
Bussolati B, Peri G, Salvidio G, Verzola D, Mantovani A, Camussi G (2003) The long pentraxin PTX3 is synthesized in IgA glomerulonephritis and activates mesangial cells. J Immunol 170:1466–1472
Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, Gingras AR, Tzima S, Vivanco F, Egido J, Tijsma O, Hack EC, Daha MR, Roos A (2003) Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol 33:465–473
Suzuki H, Moldoveanu Z, Hall S, Brown R, Julian BA, Wyatt RJ, Tomana M, Tomino Y, Mestecky J, Novak J (2007) Cytokines regulate aberrant glycosylation of IgA1 in cell lines from patients with IgA nephropathy. J Am Soc Nephrol 18:188A
Masuda M, Nakanishi K, Yoshizawa N, Iijima K, Yoshikawa N (2003) Group A streptococcal antigen in the glomeruli of children with Henoch-Schönlein nephritis. Am J Kidney Dis 41:366–370
Davin JC, Pierard G, Dechenne C, Grossman D, Nagy J, Quacoe M, Malaise M, Hall M, Jansen F, Chantraine JM, Mahieu PR (1994) Possible pathogenic role of IgE in Henoch-Schönlein purpura. Pediatr Nephrol 8:169–171
Ehara T, Shigematsu H (1998) Contribution of mast cells to the tubulointerstitial lesions in IgA nephritis. Kidney Int 54:1675–1683
Ehara T, Shigematsu H (2003) Mast cells in the kidney. Nephrology (Carlton) 8:130–138
Namgoong MK, Lim BK, Kim JS (1997) Eosinophil cationic protein in Henoch-Schönlein purpura and in IgA nephropathy. Pediatr Nephrol 11:703–706
Kawasaki Y, Hosoya M, Suzuki H (2005) Possible pathologenic role of interleukin-5 and eosino cationic protein in Henoch-Schönlein purpura nephritis. Pediatr Int 47:512–517
Chen Y, Zhou JH, Wu HS, Wang HW (2006) Role of mast cells and eosinophil cationic protein in the pathogenesis of Henoch-Schonlein purpura nephritis. Zhonghua Er Ke Za Zhi 44:407–410
Kawakami T, Yamazaki M, Mizoguchi M, Soma Y (2008) High titer of serum antiphospholipid antibody levels in adult Henoch-Schönlein purpura and cutaneous leukocytoclastic angiitis. Arthritis Rheum 59:561–567
Kawasaki Y, Imaizumi T, Matsuura H, Ohara S, Takano K, Suyama K, Hashimoto K, Nozawa R, Suzuki H, Hosoya M (2008) Renal expression of alpha-smooth muscle actin and c-Met in children with Henoch-Schönlein purpura nephritis. Pediatr Nephrol 23:913–919
Counahan R, Winterborn MH, White RH, Heaton JM, Meadow SR, Bluett NH, Swetschin H, Cameron JS, Chantler C (1977) Prognosis of Henoch-Schönlein nephritis in children. Br Med J 2:11–14
Silva GE, Costa RS, Ravinal RC, dos Reis MA, Dantas M, Coimbra TM (2008) Mast cells, TGF-beta1 and alpha-SMA expression in IgA nephropathy. Dis Markers 24:181–190
Renfrow MB, Cooper HJ, Tomana M, Kulhavy R, Hiki Y, Toma K, Emmett MR, Mestecky J, Marshall AG, Novak J (2005) Determination of aberrant O-glycosylation in the IgA1 hinge region by electron capture dissociation fourier transform-ion cyclotron resonance mass spectrometry. J Biol Chem 280:19136–19145
Acknowledgements
This work was supported in part by NIH grants DK078244, DK080301, DK082753, DK75868, DK077279, DK071802, and the Grants to the General Clinical Research Centers of the University of Alabama at Birmingham (M01 RR00032) and University of Tennessee Health Sciences Center (M01 RR00211), and by a generous gift to the University of Tennessee Pediatric Nephrology Research Support Fund by Anna and Donald Waite. The authors also acknowledge and thank all the subjects and their families involved in the research projects and all coworkers and collaborators who have participated in various aspects of these and related studies, especially including Ms. C. Barker, Ms. R. Brown, Dr. W.J. Cook, Dr. Kimberly Fisher, Dr. A. Gharavi, Ms. Sandy Grimes, Ms. S. Hall, Dr. R. Hogg, Dr. W.Q. Huang, Dr. B.A. Julian, Ms. R. Kulhavy, Dr. J.Y. Lee, Dr. R. Lifton, Dr. K. Matousovic, Dr. J. Mestecky, Dr. Z. Moldoveanu, Dr. M. Raska, Dr. M.B. Renfrow, Ms. S. Woodford, Dr. M. Tomana, Dr. Y. Tomino.
Conflict of interest
The authors have no conflict of interest to report.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found online at https://doi.org/dx.doi.org/10.1007/s00467-009-1265-z.
An erratum to this article can be found at https://doi.org/dx.doi.org/10.1007/s00467-009-1265-z
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lau, K.K., Suzuki, H., Novak, J. et al. Pathogenesis of Henoch-Schönlein purpura nephritis. Pediatr Nephrol 25, 19–26 (2010). https://doi.org/10.1007/s00467-009-1230-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-009-1230-x