
Abstract
Excessive production of reactive oxygen and nitrogen species may result in oxidative damage to tissues and organs. Oxidative stress is a pathological mechanism that contributes to the initiation and progression of liver injury. In the present study, antioxidative peptides purified from simulated gastrointestinal-digested (SGID) protein hydrolysate of Pyropia yezoensis, showed significant antioxidant activity and also showed a protective effect against acetaminophen (N-acetyl-p-aminophenol, APAP) -induced injury in HepG2 (human liver cancer cells) cells. The antioxidant activity was increased in a dose-dependent manner. Higher cell viability (73.26 ± 0.9%) and decreasing NO levels (107.6 ± 8.9%) were observed in 15 mM APAP-induced cells when treated with the concentration of (100 μg ml−1) Pyropia peptide. Py. (pep). The sequences of the eight identified peptides present in the active fractions of the protein hydrolysate included hydrophobic and aromatic amino acids, which may have been responsible for their chemoprotective and antioxidant activities. Results indicated that the treatment with the Pyropia—peptides significantly promoted the proliferation of HepG2 cells, protecting them against APAP-mediated injury, and showed a significant antioxidant capacity. This study revealed that the Py. (pep) will be beneficial in treating drug-induced oxidative stress and liver damage conditions. Py. (pep) can also serve as a better alternative for synthetic antioxidant drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acetaminophen (N-acetyl-p-aminophenol, APAP) is a commonly available antipyretic and analgesic drug that has been used extensively around the world since 1955. APAP is considered to be safe at therapeutic doses, but a single overdose can cause hepatotoxicity and liver damage [1]. There has been increasing concern over the past decade as APAP has been recognized as the major cause of acute liver failure (ALF) in adults, accounting for 30–50% of cases worldwide [2]. In the USA, APAP hepatotoxicity is responsible for 50% of overdose-related cases of ALF and 20% of liver transplant cases. Although the toxic dose of APAP that induces hepatic failure is above 150 mg/kg, there is accumulating evidence that lower doses of APAP may result in acute liver injury and liver failure [3]. Moreover, patients with COVID-19 have been widely treated with antipyretic agents, which mostly contain APAP [4]. However, these medications may be misused and subsequently increase liver injury [5].
APAP-induced hepatotoxicity and liver injury commenced by the complex mechanisms with the series of events. Formerly, cytochrome p-40 (CYP) metabolisms of the reactive metabolite covalently bind to the protein as well as diminish glutathione. Further, the loss of glutathione and the increased reactive oxygen and nitrogen species in the hepatocytes experience necrotic changes. Moreover increased oxidative stress owing to deviations in calcium homeostasis and initiation of signal transduction responses, generating the mitochondrial permeability transition. These mitochondrial permeability transition occurring along with the additional oxidative stress results in loss of mitochondrial membrane potential as well as loss of mitochondrial ATP production capability. Ultimately, loss of ATP induces necrosis. Additionally, numerous inflammatory mediators such as certain cytokines and chemokines along with these series of events can modify the toxicity [3] [6] considering this, APAP overdose causes liver failure, promoting lipid peroxidation and transcriptional activation of inflammatory mediators. In addition, APAP overdose increases ROS levels and oxidative stress.
Edible seaweeds are good sources of bioactive compounds, antioxidants, soluble dietary fiber, proteins, minerals, vitamins, phytochemicals, and polyunsaturated fatty acids [7,8,9,10]. Red, green, and brown seaweeds are rich in bioactive compounds, such as sulfated polysaccharides, phlorotannins, carotenoids, minerals, peptides, and sulfolipids [11]. Marine bioactive peptides have provided insights into the development of novel pharmaceuticals. Generally, bioactive peptides contain 3–20 amino acids and their activities are based on amino acid composition and sequence. These peptides are inactive in the parent protein sequence but can be released during gastrointestinal digestion or food processing. The biological activities of peptides mimic hormones or show drug-like activities. Bioactive peptides can alter physiological functions or have beneficial effects through binding to specific receptors and interacting with target cells or inhibiting enzymatic reactions [12].
Pyropia yezoensis, a marine red alga, is cultured and consumed mainly in China, Japan, and Korea [13]. P. yezoensis has antioxidant [14, 15], antitumor [16, 17] and anti-inflammatory activities [18, 19], and cytotoxicity [20, 21]. Glycoproteins extracted from P. yezoensis showed protective activity against APAP-induced rats [22]. Previous studies have reported the protective effects of P. yezoensis peptides in APAP-induced hepatotoxicity. Since there are very limited reports on the purification of peptides from Pyropia yezoensis by using enzymatic digestion and analyzing their protective effect. Therefore the present study was designed to purify the antioxidant peptides from P. yezoensis by various enzymatic digestion and analyze the protective effect against APAP-induced liver injury in HepG2 human liver cancer cells.
Materials and methods
Chemicals
Ammonium sulfate, trypsin, pepsin, α-chymotrypsin, fluorescein, 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AAPH), ( ±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox), 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid (ABTS), 2,2-diphenyl-1-picrylhydrazyl (DPPH), phenazine methosulfate (PMS), nicotinamide adenine dinucleotide (NADH), nitroblue tetrazolium chloride (NBT), APAP, and DAPI (4′,6-diamidino-2-phenylindole), Ascorbic acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). We purchased 2 kDa dialysis membranes from Spectrum Chemicals (Gardena, CA, USA). All of the reagents and chemicals used in this experiment were of analytical grade. Reactions were performed in deionized water.
Preparation of Pyropia yezoensis extract
P. yezoensis was obtained from Suhyup (Busan, South Korea), freeze-dried, and ground into a powder. Samples (5 g) of the powder were suspended in 100 mL ultra-pure water in 500 mL flasks, sonicated (Q500 Sonicator; QSonica Newtown, CT, USA) with 5 pulses of 5 s each with an interval of 5 s between each pulse for 1 h at 400 rpm under ice and centrifuged at 45,000 ×g for 20 min at 4 °C. The soluble protein in the supernatant was collected and precipitated with 80% ammonium sulfate. Dialysis was performed with distilled water using a 2 kDa dialysis membrane to remove the ammonium sulfate from the precipitated pellet. The soluble protein was collected, freeze-dried, and stored at 4 °C [23].
Protein hydrolysate preparation
P. yezoensis protein hydrolysates were prepared with three different enzymes and three different combinations of enzymes separately with an enzyme/substrate ratio of 1/100 (w/w) with the appropriate buffers and optimum conditions.
Six different protein hydrolysates were prepared:
-
1.
Pepsin: 10 mM glycine buffer (pH 2) at 37 °C for 2 h with rotation at 100 rpm.
-
2.
Trypsin: 50 mM sodium phosphate buffer (pH 8) at 37 °C for 4 h with rotation at 100 rpm.
-
3.
α-Chymotrypsin: 50 mM sodium phosphate buffer (pH 8) at 37 °C for 4 h with rotation at 100 rpm.
-
4.
Pepsin + Trypsin: digestion with pepsin under the optimum conditions for 2 h, adjusted to pH 8, followed by addition of trypsin and incubation for 4 h at 37 °C with rotation at 100 rpm.
-
5.
Pepsin + α-chymotrypsin: digestion with pepsin under the optimum conditions for 2 h, adjusted to pH 8, followed by addition of α-chymotrypsin and incubation for 4 h at 37 °C with rotation at 100 rpm.
-
6.
Pepsin + Trypsin + α-chymotrypsin: digestion with pepsin under the optimum conditions for 2 h, adjusted to pH 8, followed by addition of trypsin and α-chymotrypsin and incubation for 4 h at 37 °C with rotation at 100 rpm.
After enzyme digestion, the enzymes were deactivated by heating for 10 min at 100 °C. The hydrolysates were centrifuged at 10,000 ×g (4 °C) for 30 min and the supernatants were stored for further study.
Antioxidant activity
ORAC assay
The oxygen radical absorbance capacity (ORAC) assay of Pyropia protein hydrolysates was measured using the fluorescent probe fluorescein (FL). An automated ORAC assay was carried out on a Synergy HTX multi-mode microplate reader with fluorescence filters at an excitation wavelength of 485 nm and emission wavelength of 535 nm. The measurements were made in plates with 96 black-bottomed wells. The reaction was performed at 37 °C with the thermal decomposition of AAPH [24].
A stock solution of the fluorescent probe was prepared by dissolving 44 mg FL in 100 mL phosphate-buffered saline (PBS, pH 7.0, final concentration 75 mM), and storing at 4 °C in the dark. A 78 nM working solution was prepared daily by dilution of 0.167 mL of the stock solution in 25 mL phosphate buffer. The AAPH radical (221 mM) was prepared daily by adding 600 mg AAPH to 10 mL PBS. The reference standard used was 20 μM Trolox solution that was prepared daily in PBS from a 1 mM stock solution kept in the freezer at − 20 °C. As the ORAC assay is extremely sensitive, the samples were diluted before analysis to avoid interference. To each well was added 150 µL FL (78 nM) and 25 µL the sample, blank (PBS), or standard (Trolox, 20 μM), and then 25 µL AAPH (221 mM) was added. Variation in measurements between wells can arise because of the low conductivity of the polypropylene plates. To avoid this problem, the plates were heated to 37 °C for 15 min prior to adding AAPH. The fluorescence was measured immediately after adding AAPH and then the measurements were taken in triplicate every 2 min until 60 min. The ORAC values, expressed as mM Trolox equivalents (mM TE/mg), were calculated by applying the following Eq. 1.
where CTrolox is the concentration (μM) of Trolox (20 μM), k is the sample dilution factor, and AUCSample, AUCBlank, and AUCTrolox are the areas under the fluorescence decay curves of the sample, blank, and Trolox, respectively.
TEAC
Trolox equivalent antioxidant capacity (TEAC) was determined as described by [25] with minor modifications based on the capacity of the sample to inhibit the ABTS radical (ABTS+) compared to the reference antioxidant standard Trolox. The ABTS+ radical was generated by a chemical reaction with potassium persulfate (K2S2O8). For this purpose, 25 mL ABTS (7 mM) was spiked with 440 µL K2S2O8 (140 mM) and allowed to stand in the dark at room temperature for 12–16 h. The working solution was prepared by diluting aliquots of the previous solution in PBS (pH 7.4) until the absorbance at 734 nm (A734) was 0.70 ± 0.02. The diluted ABTS working solution 200 μL + sample or PBS or Trolox standard 10 μL was added, incubated for 5 min in the dark, and the A734 was determined using a multi-mode microplate reader (Synergy HTX; BioTek, Winooski, VT, USA) [24]. A Trolox calibration curve was also prepared for the concentration range of 0–400 μM, and the inhibitory effect was interpolated to calculate the concentration in Trolox equivalents (mM TE/mg). All determinations were carried out in triplicate.
DPPH radical scavenging activity
The DPPH radical scavenging activity of protein hydrolysates of P. yezoensis was determined as described previously [26], with minor modifications. Briefly, 0.4 mM DPPH solution (750 μL) solubilized in ethanol was added to 150 µL protein hydrolysate. The mixture was vortexed and incubated at 25 °C for 30 min in the dark. The absorbance at 517 nm (A517) of the mixture was measured, ascorbic acid was used as standard and the DPPH radical scavenging activity was calculated by using Eq. 2.
where A0 is the absorbance of the control and A1 is the absorbance of the sample.
Superoxide radical scavenging activity
The superoxide anion scavenging activity was measured based on a method described previously [27]. Superoxide radicals were generated in a PMS-NADH system by oxidation of NADH and assayed by reduction of NBT. In this experiment, the superoxide radicals were generated in sodium phosphate buffer (100 mM, pH 7.4) containing 50 µL NBT (150 μM) solution, 50 µL NADH (468 μM) solution, and 50 µL Pyropia protein hydrolysate (1 mg/mL). The reaction was started by adding 50 µL PMS solution (60 μM). The reaction mixture was incubated at 25 °C for 5 min, and the absorbance at 560 nm (A560) was measured against the corresponding blank solution. The decrease in the extent of NBT reduction, measured according to the absorbance of the reaction mixture, was correlated with the superoxide radical scavenging activity of the protein hydrolysates, ascorbic acid was used as standard. The percentage of superoxide radical scavenging was calculated using the following Eq. 3.
where A0 is the absorbance of the control and A1 is the absorbance of the sample.
Hydroxyl radical scavenging
The hydroxyl radical scavenging activity was measured using the Fenton reaction according to [28]. Briefly, the reaction mixture contained 60 µL 1.0 mM FeCl2, 90 µL 1 mM 1,10-phenanthroline, 2.4 mL 0.2 M phosphate buffer (pH 7.8), 150 µL 0.17 M H2O2, and 1.5 mL Pyropia protein hydrolysate. The reaction was started by adding H2O2. After incubation at room temperature for 5 min, the A560 was measured with a spectrophotometer, ascorbic acid was used as standard. The hydroxyl radical scavenging activity was calculated using the following Eq. 4.
Molecular weight cutoff separation
The Pyropia protein hydrolysate was separated using an ultrafiltration membrane (Vivaflow 200 Laboratory Cross Flow Cassette; Sartorius, Goettingen, Germany).
The protein hydrolysate was separated into three different molecular weight fractions:
-
1.
> 10 kDa
-
2.
5–10 kDa
-
3.
3–5 kDa
-
4.
< 3 kDa
The antioxidant activity was analyzed for the above four fractions, among which the < 3 kDa fraction showed the greatest antioxidant activity. The < 3 kDa fraction was therefore further purified by anion exchange chromatography.
Fast protein liquid chromatography (FPLC)
The obtained < 3 kDa hydrolysate was further purified by fast protein liquid chromatography (FPLC) (AKTA Prime Plus; GE Healthcare, Piscataway, NJ, USA) on a HiPrep 16/10 diethyl aminoethyl fast flow (DEAE FF) ion exchange column. The freeze-dried < 3 kDa protein hydrolysate was dissolved at a concentration of 20 mg/mL in 20 mM Tris–HCl (pH 9). Aliquots of 5 mL were loaded onto a HiPrep 16/10 DEAE FF ion exchange column equilibrated with 20 Mm Tris–HCl (pH 9.5), and eluted with a linear gradient of NaCl (0–1 M) in the same buffer at a flow rate of 2 mL/min. Each fraction (5 mL) was monitored at 214 nm, and fractions corresponding to the peaks were collected and pooled. The pooled fractions were freeze-dried and the antioxidant and hydroxyl radical scavenging activities were analyzed. The fraction with the highest antioxidant and hydroxyl radical scavenging activities was further purified by high-performance liquid chromatography (HPLC), to obtain a pure peptide.
Preparative RP-HPLC
The activity-rich fraction was further purified by reversed-phase HPLC (RP-HPLC) using a Luna® 5 µm C18(2) 100 Å, LC Column (250 × 10 mm; Phenomenex, Torrance, CA, USA), attached to a Dionex Chromeleon 7.2 Chromatography Data System (Thermo Scientific, Waltham, MA, USA). Elution was performed with solution A (0.1% formic acid in deionized water) and solution B (0.1% formic acid in acetonitrile) eluted with a linear gradient of acetonitrile (0–5 min 90% of solution A and 10% solution B, 5–55 min 20% solution A, and 80% solution B, 55–60 min 20% solution A and 80% solution B, 60–66 min 90% solution A and 10% solution B) at a flow rate of 1 mL/min. The absorbance was monitored at 214 nm (A214). Ascorbic acid was used as a standard to compare the results.
Determination of peptide sequence by LC–MS/MS
The amino acid sequence was confirmed by de novo sequencing of peptides from proteolytic fragments or peptide extracts by liquid chromatography with tandem mass spectrometry (LC-MS-MS).
Mass spectrometry was performed using a Micro Q-TOF III mass spectrometer (Bruker Daltonics, Billerica, MA, USA). Filtered (mini start syringe filter, pore size 0.45 μm; Satorius) samples were injected in a volume of 10 μL through a Poroshell 120 EC-C18 (2.1 × 100 mm, 2.7 μm; Agilent Technologies, Santa Clara, CA, USA) LC column and separated using an UltiMate 3000 system (Dionex, Sunnyvale, CA, USA). Chromatographic separation was achieved at a flow rate of 0.2 mL/min using a gradient elution program, starting with 98% Solvent AA (H2O/FA = 100/0.1, v/v) and 2% solvent B (acetonitrile/FA = 100/0.1, v/v) up to 2 min. This was further changed gradually to 70% A and 30% B at 20 min. Later, the gradient was changed rapidly to 5% A and 95% B from 20 to 21 min and maintained up to 26 min. Further, the program rapidly changed to 98% eluent A and 2% B at 27 min and was maintained up to 35 min.
Mass spectrometry was performed with a Micro Q-TOF III mass spectrometer (Bruker Daltonics). Samples were introduced to MS via ESI + using the following MS parameters: Source: capillary voltage, 3500; nebulizer, 0.8; dry gas, 5.5; dry temp., 180 °C. Transfer: funnel 1RF, 400; funnel 2RF, 400; ISCID energy, 0 eV; hexapole RF, 250. ion energy, 5.0 eV; low mass, mass/charge ratio (m/z) 300; collision cell with collision energy, 7 eV; collision RF, 600; transfer time, 80 μs; pre-pulse storage, 10 μs. Further, the collected spectra were scanned with m/z 50–2000. MS spectra were generated by collision-induced dissociation of the metabolite ions at a normalized collision energy of 100 Vpp.
The peptides De novo sequencing analyzes the amino acid sequence based on the analyzed MS/MS spectrum. Eight peptide amino acid sequences were identified.
Cell culture
HepG2 Human liver cancer cells (cat. no. HB-8065) were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured at 37 °C with 5% CO2 in minimum essential medium (MEM; Sigma–Aldrich; Merck KGaA) supplemented with 10% FBS (GenDEPOT Inc., Barker, TX, USA) containing 100 U/ml penicillin, and 100 mg/ml streptomycin. The medium was replaced every 2 days.
Cell viability assay
Cell viability was estimated using a Cyto X Cell Viability Assay kit (Cat. No. EZ-1000; EZ-Cytox, Oxford, UK). Cells were seeded in 96-well plates at 2 × 104 cells/well in 100 µL medium and allowed to attach for 24 h at 37 °C. Then the attached cells were treated with Py. pep (5, 25, 50, or 100 µg/mL) and 15 mM APAP (A7085; Sigma-Aldrich-Merck KGaA) in serum-free MEM (SFM) for 18 h at 37 °C [29]. Cyto X solution was added to the cells, followed by incubation for 1 h at 37 °C and the absorbance at 450 nm (A450) was measured using a FilterMAX F5 microplate reader (Molecular Devices, Downingtown, PA, USA). Morphological changes in the cells were subsequently examined using a light microscope (magnification, × 200; Eclipse TS100-F; Nikon, Tokyo, Japan).
DAPI staining assay
The cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde. The fixed cells were incubated at 37 °C for 20 min and washed twice with PBS. Then the cells were stained with 1 µg/mL DAPI and incubated at room temperature for 20 min in the dark. The stained cells were observed under a fluorescence microscope (ECLIPSE TS100-F; Nikon) [30].
Nitric oxide (NO) assay
The nitrite concentration in the culture medium was determined spectrophotometrically as described previously [19]. Briefly, cells were seeded in 48-well plates at 2 × 106 cells/well and incubated for 24 h at 37 °C. The cells were treated with Py. pep (5, 25, 50, and 100 µg/mL) and 15 mM APAP in SFM for 18 h at 37 °C. Subsequently, 90 µL culture medium was transferred into each well of 96-well plates, and 50 µL Griess reagent (G4410; Sigma-Aldrich-Merck KGaA) was added. The plates were incubated for 10 min at 37 °C, after which the absorbance at 540 nm (A540) was measured using a FilterMAX F5 microplate reader.
Statistical analysis
Results are presented as the mean ± SD of three independent experiments. The significance of differences among multiple means was assessed via one-way or two-way ANOVA followed by Dunnett’s multiple comparisons test using GraphPad Prism software (version 9; GraphPad Software, San Diego, CA, USA). In all analyses, P < 0.05 was taken to indicate statistical significance.
Results and discussion
Protein extraction and purification
Protein was extracted from the marine alga P. yezoensis via 80% ammonium sulfate precipitation and desalted using a 2 kDa dialysis membrane. The protein concentration was measured using the Lowry method [31] and was about 40% of the total biomass dry weight. This is consistent with the protein content of 41.4% dry weight in Pyropia reported previously [32], which is three times higher than other seaweeds. Moreover, several studies have shown that Pyropia has protein contents ranging from 24.6% to 47.4% dry weight when grown under different conditions [11] [33,34,35]. The SDS-PAGE profile of the proteins extracted from P. yezoensis is presented in Fig. 1a. The relatively intense bands at 29–33 and 18–22 kDa were likely to correspond to phycobiliproteins (e.g., phycoerythrin), which are typical pigment proteins related to photosynthesis in red seaweed species. Phycobiliprotein from P. yezoensis has previously been reported [23]. Figure 1b shows the glycoproteins present in P. yezoensis.

The purified pyropia protein was analyzed in 12.5% SDS-PAGE. a Polyacrylamide gel stained with Coomassie brilliant blue R-250, M: molecular weight marker, Lane 1–10 μg, 2–20 µg, 3–30 µg Pyropia protein. b Polyacrylamide gel stained with glycoprotein stain with M: molecular weight marker. Lane 1–10 μg, 2–20 µg, 3–30 µg Pyropia protein. Glycoprotein protein bands appeared approximately 75 ~ 50 and 20 ~ 15 kDa
Antioxidant activity of P. yezoensis protein hydrolysate
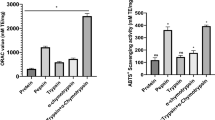
Macroalgae, particularly species with high protein contents, are also of significant interest as sources of bioactive peptides. Most of the proteins are subjected to hydrolysis to release the peptides. The majority of bioactive peptides are reported after hydrolysis by gastrointestinal enzymes or proteolytic or peptidolytic enzymes [36]. In the present study, the purified protein was digested with six different combinations of enzymes: pepsin, trypsin, α-chymotrypsin, pepsin + trypsin, pepsin + α-chymotrypsin, pepsin + trypsin + α-chymotrypsin. Protein hydrolysates obtained by digestion with these different enzyme combinations were analyzed for their antioxidant activities, i.e., oxygen radical absorbance capacity (ORAC), Trolox equivalent antioxidant capacity (TEAC), and 2,2-diphenyl-1-picrylhydrazyl (DPPH), superoxide radical, and hydroxyl radical scavenging activities. Among the six different protein hydrolysates, the simulated gastrointestinal digested (SGID) hydrolysate obtained with the combination of three enzymes (pepsin + trypsin + α-chymotrypsin) showed significant ORAC and TEAC antioxidant activities compared to other protein hydrolysates, and unhydrolyzed Pyropia protein. The ORAC value of unhydrolyzed Pyropia protein was 106.03 ± 7.86 mM TE/mg, while the SGID protein hydrolysate showed approximately four times higher ORAC activity of 432.763 ± 23.04 mM TE/mg (Fig. 2a).

Antioxidant activity of Pyropia protein and six different protein hydrolysates of Pyropia a ORAC activity, b TEAC activity, c DPPH radical scavenging activity, d Superoxide radical scavenging activity, e Hydroxyl radical scavenging activity. Values are each expressed as a mean ± SD (n = 3). Results are presented as means ± SD, different alphabets denote significant differences (p < 0.05)
The ABTS scavenging activity of unhydrolyzed Pyropia protein was 12.44 ± 3.2 mM TE/mg, while that of the SGID hydrolysate was approximately five times higher with a value of 64.42 ± 3.08 mM TE/mg, which was also the highest value among the hydrolysates examined (Fig. 2b). The SGID hydrolysate also showed higher antioxidant activities than the other protein hydrolysates and unhydrolyzed Pyropia protein. SGID and unhydrolyzed Pyropia protein showed DPPH radical scavenging activities of 58.99% ± 3.28% and 10.86% ± 2.11%, respectively, superoxide radical scavenging activities of 47.65% ± 1.86% and 7.8% ± 1.47%, respectively, and hydroxyl radical scavenging activities of 61.63% ± 2.45% and 8.3% ± 1.1%, respectively Fig. 2c–e. These results suggest that proteolysis may have promoted the release of other compounds, such as polyphenols, with antioxidant activity that may have contributed to the overall higher ORAC values of Pyropia protein hydrolysate in a synergistic manner. Increases in antioxidant activity in SGID have also been reported for whey protein α-lactalbumin, which showed a considerable increase in ORAC value compared to the unhydrolyzed control [37]. A pepsin hydrolysate obtained from Nori sheets (Porphyra) also showed an increase in the DPPH scavenging ability up to 60% following proteolytic treatment compared to 30% by the unhydrolyzed algal extract [38].
Beaulieu et al. [39] compared the antioxidant activities of both protein extracts and α-chymotrypsin hydrolysates of Palmaria palmata, and showed that hydrolysis improved the antioxidant activity by up to sixfold in FRAP and ORAC assays. [40] reported the digestion resistance of bioactive peptides from a hydrolysate of Undaria pinnatifida against gastrointestinal proteases (pepsin, trypsin, and chymotrypsin), while their cardioprotective properties were unaffected. It has also been reported that peptides derived from P. palmata retain antioxidant and dipeptidyl peptidase-IV (DPP-IV) inhibitory activities after simulated gastric digestion [41, 42].
Antioxidant activities of different molecular weight fractions
The SGID Pyropia protein hydrolysate was further separated into four different fractions based on molecular weight, i.e., > 10 kDa, 10–5 kDa, 5–3 kDa, < 3 kDa, using molecular weight cutoff membranes (Viva Flow 200; Sartorius, Goettingen, Germany). The four different molecular weight fractions were analyzed for antioxidant activities. The low-molecular-weight ~ 3 kDa fraction showed the highest antioxidant activity among the fractions in all assays performed (Fig. 3a–e). Similar results were reported in a previous study indicating that α-chymotrypsin hydrolysate of P. palmata fractions with molecular weights below 10 kDa had higher activity than those with molecular weights above 10 kDa [39]. Our results agree with previous reports that peptides exhibiting antioxidant activity generally have low molecular weights [43]. Many studies have reported potential health benefits of low-molecular-weight peptides isolated from marine algae [44].

Antioxidant activity of different molecular weight fractions of pyropia protein hydrolysates a ORAC activity, b TEAC activity, c DPPH radical scavenging activity, d Superoxide radical scavenging activity, e Hydroxyl radical scavenging activity. Values are each expressed as a mean ± SD (n = 3). Results are presented as means ± SD, different alphabets denote significant differences (p < 0.05)
Purification and antioxidant activity of ~ 3 kDa) molecular weight fraction
The antioxidant activity-rich (~ 3 kDa) fraction was further purified by anion exchange chromatography using FPLC. Gradient elution was performed in 1 M NaCl. In all, eight peaks were obtained, including those of non-binding peptides (Fig. 4). The eight peaks were collected and analyzed separately for their antioxidant activities. Among them, fraction 4 showed the highest antioxidant activity (Fig. 5).

Fast protein liquid chromatography (FPLC) profile of 3 ~ kDa peptide. 5 ml loaded onto a HiPrepTM 16/10 DEAE FF anion-exchange column. Separation was performed at 2 mL min– 1. Elution was monitored at 214 nm

Antioxidant activity of Fraction collected from FPLC a ORAC value of fractions, b ABTS activity of fraction. Values are each expressed as a mean ± SD (n = 3). Results are presented as means ± SD, different alphabets denote significant differences (p < 0.05)
Purification of the activity-rich fraction 4
Fraction 4 was further purified by RP-HPLC. The HPLC profile of fraction 4 showed eight major peaks (Fig. 6).

RP-HPLC using Luna® 5 µm C18(2) 100 Å, LC Column 250 × 10 mm (Phenomenex., CA, USA), attached to Dionex system (Thermo Scientific™ Dionex™ Chromeleon™ 7.2 Chromatography, Waltham, Massachusetts, USA.,). Elution was performed with solution A (0.1% formic acid in deionized water) and solution B (0.1% formic acid in acetonitrile) eluted with a linear gradient of acetonitrile (0–80% in 0–66 min) at a flow rate of 1 mL/min. a HPLC profile of Fraction 4. b HPLC profile of ascorbic acid
Peptide identification by MS/MS
MS/MS identified eight peptides, as shown in Fig. 7a–h and Table 1.


MS/MS spectra of identified peptides (a–h) and sequences of peptides 1–8
The peptides were further identified by MS/MS. In the MS/MS result there are 8 peptides were identified. They are presented in Fig. 7a–h and Table 1.
Eight peptides that were responsible for the antioxidant activities were identified by HPLC. Sequencing of these peptides suggested that the antioxidant activity was reinforced not only by the low-molecular-weight peptides, but also by the presence of hydrophobic and/or aromatic amino acids in the sequences generated after protein hydrolysis. In fact, the presence of hydrophobic and specific aromatic residues (i.e., phenylalanine, tyrosine, leucine, and valine) along with histidine has been reported as a specific feature of some bioactive peptides with antioxidant activity [45]. In the present study, the obtained peptide sequences also contained hydrophobic and aromatic residues, which may explain the observed antioxidant activity.
In a previous study, a peptide isolated from Pyropia was composed of Thr-Tyr-Ile-Ala and had the highest ACE inhibitory activity (IC50: 197.5 ± 47.7 μM) followed by Tyr-Leu-Val-Ala (IC50: 259.7 ± 34.0 μM) and Asp-Tyr-Tyr-Lys-Arg (IC50: 628.9 ± 24.9 μM). These three peptides have Tyr within their sequence. Furthermore, peptides with Tyr within their sequence at the C3 and C4 positions show high antioxidant activity. The presence of Tyr in such positions is a structural feature associated with high ORAC activity [46]. In the present study, the obtained peptide sequences Val-Ser-Tyr and His-Pro-Asp-Tyr had a Tyr residue at the C3 and C4 positions, respectively, which may have been responsible for their excellent ORAC activity.
Cermeño et al. [46] also fractionated P. dioica protein hydrolysate and selected the most bioactive fraction for peptide identification. Tyr-Leu-Val-Ala, present in C-phycocyanin, showed multifunctional activity, with high antioxidant, ACE, and DPP-IV inhibitory activity. After SGID, Tyr-Leu-Val-Ala retained its ORAC and ACE inhibitory activities. Asp-Tyr-Tyr-Lys-Arg, present in C-allophycocyanin, had the highest ORAC activity. Asp-Tyr-Tyr-Lys-Arg was extensively degraded on incubation with digestive enzymes, but its ORAC activity was significantly enhanced after SGID. Finally, Thr-Tyr-Ile-Ala, present in β-phycoerythrin, had the highest ACE inhibitory activity both before and after SGID. Based on the results presented here, the peptides identified from P. yezoensis protein hydrolysate may be useful as multifunctional components in the pharmaceutical and food industries.
Effects of Pyropia polypeptide fraction on APAP-induced toxicity in HepG2 cells
Cell viability
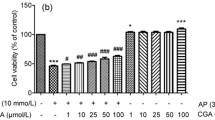
The effects of the Py. peptide fraction on the viability of APAP-treated HepG2 liver cells were analyzed. The previous studies showed that the HepG2 cells treated with 15 mM APAP for 18 h showed 60% cells viability [29, 47] based on that, in the present study the HepG2 cells treated with 15 mM APAP for 18 h showed 60.97% ± 2.27% viability compared to untreated controls (Fig. 8a). Treatment of 15 mM APAP-treated HepG2 cells with 5, 25, 50, and 100 μg/mL Py. (pep) showed dose-dependent increases in cell viability (64.02% ± 1.4%, 70.77% ± 3.04%, 72.17% ± 3.19%, and 73.26% ± 0.9%, respectively) compared to controls treated with 15 mM APAP alone. The cell viability was increased in the dose-dependent manner. Previous study with the recombinant protein isolated from Pyropia yezoensis has also increased the cell viability in the APAP-induced HepG2 cells in a dose-dependent manner [48]. Additionally, a study by [23] discovered the prominent cell viability in APAP-induced HepG2 cells when treated with Py. (pep) in a dose-dependent manner. Photomicrographs also elucidated the increased cell viability when treated with the increasing Py. (pep) concentration (Fig. 8b).

a Protective effects of Py. (pep) on acetaminophen (APAP)-induced liver damage in HepG2 cells. Cell viability was measured by Cyto X assays, and the results are presented as percentages of surviving cells compared with the control (non‑treated group). Data are presented as the mean ± SD of three independent experiments and were analyzed using two‑way ANOVA. *p < 0.05 vs. control group; #p < 0.05 vs. the 15 mM APAP group. b Morphological changes in cells following treatment with APAP alone or APAP + Py. (pep). Upper and lower panels show bright field and fluorescence images. Scale bar, 100 μm
Py. (pep) decreases APAP‑induced oxidative stress in HepG2 cells
Oxidative stress is involved in APAP-induced liver failure, and liver tissue is damaged by various cytokines and high levels of NO following an APAP overdose [49]. Griess reagent was used to investigate NO production in HepG2 cells treated with APAP with or without Py. (pep). The APAP group showed a significantly elevated NO level of 128.7% ± 4.1% compared to the control group, whereas cotreatment with Py. (pep) significantly suppressed the increase in NO level in a concentration-dependent manner, with the 100 μg/mL Py. (pep) group showing a NO level of 107.6% ± 8.9% to the control group (Fig. 9). Hence these results indicated the effective role of Py. (pep) on subsiding NO levels in the APAP-induced liver cells. A similar study on the APAP induced HepG2 cells treated with Py. (pep) with dose-dependent manner also exhibited the reduction of NO levels [22].

Py. (pep) treatment restores APAP‑induced oxidative stress in APAP‑induced HepG2 cells. The liver cells were incubated with 15 mM APAP with or without various concentrations of Py. (pep) for 18 h. NO levels were analyzed using a Griess assay. Data are presented as the mean ± SD of three independent experiments and were subjected to two‑way ANOVA. *p < 0.05 vs. the control group; #p < 0.05 vs. the 15 mM APAP group
Previous studies on seaweeds and APAP-induced hepatotoxicity have focused on Sargassum species (Hizikia fusiformis syn. Sargassum fusiforme), red algae (P. yezoensis), green algae (Ulva reticulata and Chlorella sorokiniana), and sulfated polysaccharides (fucoidan) [21, 50]. In a previous study, prevention of APAP-induced hepatotoxicity was associated with a 14 kDa protein of P. yezoensis designated PYP [21].
Although APAP is an over-the-counter drug that is widely used for analgesic and antipyretic purposes, an overdose can cause liver injury, liver failure, and even death [51]. Therefore, a number of studies have investigated APAP metabolism in the liver and various factors with protective effects against APAP-induced liver toxicity. There have been few studies of the mechanisms of action of Pyropia peptides since [22] Hwang et al. reported the effects of PYP against APAP-induced liver cell injury.
In the present study, we investigated the chemoprotective activities of Py. (pep) against APAP-induced cytotoxicity. Py. (pep) exhibited strong chemoprotective activities against APAP-induced toxicity and thus had protective effects against liver cell injury. The present study revealed that Py. (pep) at 0–100 µg/ml was non-toxic in HepG2 cells and reversed the effects of APAP-induced hepatotoxicity.
Conclusion
This study concluded that the unhydrolyzed protein from the Pyropia yezoensis showed the least antioxidant activity compared to enzyme-digested protein hydrolysates of Pyropia yezoensis. Further, the low molecular weight peptide fractions derived using molecular weight cut-off membranes exhibited significant antioxidant activity. However, the combination of peptides identified and purified from these protein hydrolysates has shown remarkable antioxidant activity as well as productive effects (increasing cell viability and reducing oxidative stress) against the APAP-induced HepG2 liver cells. APAP overdose increased apoptosis, whereas co-treatment with Py. (pep) resulted in a dose-dependent decrease in apoptosis. These combined peptides consist of 8 different peptides with hydrophobic, aromatic amino acids, and Tyrosine in their sequences which could be facilitating antioxidant and chemoprotective activity. Further synthesizing and in-vivo studies of these peptides will be valuable in the food and pharmaceutical industry.
References
Davidson DG, Eastham WN (1966) Acute liver necrosis following overdose of paracetamol. Br Med J 27:497–499. https://doi.org/10.1136/bmj.2.5512.497
Bunchorntavakul C, Reddy KR (2013) Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther 37:3–17. https://doi.org/10.1111/apt.12109
Yoon E, Babar A, Choudhary M et al (2016) Acetaminophen-induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol 4:131–142. https://doi.org/10.14218/JCTH.2015.00052
Feng G, Zheng KI, Yan QQ et al (2020) COVID-19 and liver dysfunction: current insights and emergent therapeutic strategies. J Clin Transl Hepatol 8:18–24
Wang J, Zhang L, Shi Q et al (2022) Targeting innate immune responses to attenuate acetaminophen-induced hepatotoxicity. Biochem Pharmacol 202:115142. https://doi.org/10.1016/j.bcp.2022.115142
Ramachandran A, Jaeschke H (2017) Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res 3:157–169. https://doi.org/10.18053/jctres.03.2017S1.002
Ulagesan S, Nam TJ, Choi YH (2021) Biogenic preparation and characterization of Pyropia yezoensis silver nanoparticles (P.y AgNPs) and their antibacterial activity against Pseudomonas aeruginosa. Bioprocess Biosyst Eng 44:443–452. https://doi.org/10.1007/s00449-020-02454-x
Ulagesan S, Choi J-W, Nam T-J et al (2020) Peptidyl-prolyl isomerase and the biological activities of recombinant protein cyclophilin from Pyropia yezoensis (PyCyp). Protein Expr Purif 172:105636. https://doi.org/10.1016/j.pep.2020.105636
Bharathiraja S, Seo H, Manivasagan P et al (2016) In vitro photodynamic effect of phycocyanin against breast cancer cells. Molecules 21(11):1470. https://doi.org/10.3390/molecules21111470
Bharathiraja S, Manivasagan P, Oh Y-O et al (2017) Astaxanthin conjugated polypyrrole nanoparticles as a multimodal agent for photo-based therapy and imaging. Int J Pharm 517:216–225
Cian RE, Fajardo MA, Alaiz M et al (2014) Chemical composition, nutritional and antioxidant properties of the red edible seaweed Porphyra columbina. Int J Food Sci Nutr 65:299–305. https://doi.org/10.3109/09637486.2013.854746
Pangestuti R, Kim SK (2017) Bioactive peptide of marine origin for the prevention and treatment of non-communicable diseases. Mar Drugs 15:1–23. https://doi.org/10.3390/md15030067
Niwa K (2010) Genetic analysis of artificial green and red mutants of Porphyra yezoensis Ueda (Bangiales, Rhodophyta). Aquaculture 308:6–12. https://doi.org/10.1016/j.aquaculture.2010.08.007
Ma X-T, Sun X-Y, Yu K et al (2017) Effect of content of sulfate groups in seaweed polysaccharides on antioxidant activity and repair effect of subcellular organelles in injured HK-2 cells. Oxid Med Cell Longev 2017:2542950. https://doi.org/10.1155/2017/2542950
Toyosaki T, Iwabuchi M (2009) New antioxidant protein in seaweed (Porphyra yezoensis Ueda). Int J Food Sci Nutr 60:46–56. https://doi.org/10.1080/09637480802345591
Yu X, Zhou C, Yang H et al (2015) Effect of ultrasonic treatment on the degradation and inhibition cancer cell lines of polysaccharides from Porphyra yezoensis. Carbohydr Polym 117:650–656. https://doi.org/10.1016/j.carbpol.2014.09.086
Park SJ, Ryu J, Kim IH et al (2015) Activation of the mTOR signaling pathway in breast cancer MCF-7 cells by a peptide derived from Porphyra yezoensis. Oncol Rep 33:19–24. https://doi.org/10.3892/or.2014.3557
Yanagido A, Ueno M, Jiang Z et al (2018) Increase in anti-inflammatory activities of radical-degraded porphyrans isolated from discolored nori (Pyropia yezoensis). Int J Biol Macromol 117:78–86. https://doi.org/10.1016/j.ijbiomac.2018.05.146
Lee HA, Kim IH, Nam TJ (2015) Bioactive peptide from Pyropia yezoensis and its anti-inflammatory activities. Int J Mol Med 36:1701–1706. https://doi.org/10.3892/ijmm.2015.2386
Choi JW, Kim IH, Kim YM et al (2016) Pyropia yezoensis glycoprotein regulates antioxidant status and prevents hepatotoxicity in a rat model of D-galactosamine/lipopolysaccharide-induced acute liver failure. Mol Med Rep 13:3110–3114. https://doi.org/10.3892/mmr.2016.4932
Hwang H-J, Kwon M-J, Kim I-H, Nam T-J (2008) Chemoprotective effects of a protein from the red algae Porphyra yezoensis on acetaminophen-induced liver injury in rats. Phyther Res. 22:1149–1153. https://doi.org/10.1002/ptr.2368
Kim I-H, Choi J-W, Nam T-J (2020) PYP1-4 peptide from Pyropia yezoensis protects against acetaminophen-induced hepatotoxicity in HepG2 cells. Exp Ther Med 19:849–860. https://doi.org/10.3892/etm.2019.8304
Ulagesan S, Nam TJ, Choi YH (2021) Extraction and purification of R-phycoerythrin alpha subunit from the marine red algae pyropia yezoensis and its biological activities. Molecules. https://doi.org/10.3390/molecules26216479
Zulueta A, Esteve MJ, Frígola A (2009) ORAC and TEAC assays comparison to measure the antioxidant capacity of food products. Food Chem 114:310–316. https://doi.org/10.1016/j.foodchem.2008.09.033
Re R, Pellegrini N, Proteggente A et al (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26:1231–1237. https://doi.org/10.1016/s0891-5849(98)00315-3
Yang B, Zhao M, Shi J et al (2008) Effect of ultrasonic treatment on the recovery and DPPH radical scavenging activity of polysaccharides from longan fruit pericarp. Food Chem 106:685–690. https://doi.org/10.1016/j.foodchem.2007.06.031
Fontana M, Mosca L, Rosei MA (2001) Interaction of enkephalins with oxyradicals. Biochem Pharmacol 61:1253–1257. https://doi.org/10.1016/S0006-2952(01)00565-2
Wenli Y, Yaping Z, Bo S (2004) The radical scavenging activities of radix puerariae isoflavonoids: a chemiluminescence study. Food Chem 86:525–529. https://doi.org/10.1016/j.foodchem.2003.09.005
Kim I, Choi J, Nam T (2019) PYP1-4 peptide from Pyropia yezoensis protects against acetaminophen-induced hepatotoxicity in HepG2 cells. Exp Ther Med. https://doi.org/10.3892/etm.2019.8304
Chazotte B (2011) Labeling nuclear DNA using DAPI. Cold Spring Harb Protoc 6:80–83. https://doi.org/10.1101/pdb.prot5556
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. https://doi.org/10.1016/s0021-9258(19)52451-6
Cho TJ, Rhee MS (2020) Health functionality and quality control of laver (porphyra, pyropia): current issues and future perspectives as an edible seaweed. Mar Drugs 18:1–31. https://doi.org/10.3390/md18010014
Dawczynski C, Schubert R, Jahreis G (2007) Amino acids, fatty acids, and dietary fibre in edible seaweed products. Food Chem 103:891–899. https://doi.org/10.1016/j.foodchem.2006.09.041
Smitha JL, Summers G, Wong R (2010) Nutrient and heavy metal content of edible seaweeds in New Zealand. New Zeal J Crop Hortic Sci 38:19–28. https://doi.org/10.1080/01140671003619290
Pimentel FB, Cermeño M, Kleekayai T et al (2020) Effect of in vitro simulated gastrointestinal digestion on the antioxidant activity of the red seaweed Porphyra dioica. Food Res Int 136:109309. https://doi.org/10.1016/j.foodres.2020.109309
Sharma S, Singh R, Rana S (2011) Bioactive peptides: a review. Int J Bioautomation 15:223–250. https://doi.org/10.1093/fqs/fyx006
Báez J, Fernández-Fernández AM, Tironi V et al (2021) Identification and characterization of antioxidant peptides obtained from the bioaccessible fraction of α-lactalbumin hydrolysate. J Food Sci 86:4479–4490. https://doi.org/10.1111/1750-3841.15918
Parimelazhagan I, Mehta A (2017) Changes in the antioxidant potential of nori sheets during in vitro digestion with pepsin. J Aquat Food Prod Technol 26:163–171. https://doi.org/10.1080/10498850.2015.1125981
Beaulieu L, Sirois M, Tamigneaux É (2016) Evaluation of the in vitro biological activity of protein hydrolysates of the edible red alga, Palmaria palmata (dulse) harvested from the Gaspe coast and cultivated in tanks. J Appl Phycol 28:3101–3115. https://doi.org/10.1007/s10811-016-0850-3
Sato M, Hosokawa T, Yamaguchi T et al (2002) Angiotensin I-converting enzyme inhibitory peptides derived from wakame (Undaria pinnatifida) and their antihypertensive effect in spontaneously hypertensive rats. J Agric Food Chem 50:6245–6252. https://doi.org/10.1021/jf020482t
Harnedy PA, O’Keeffe MB, Fitzgerald RJ (2015) Purification and identification of dipeptidyl peptidase (DPP) IV inhibitory peptides from the macroalga Palmaria palmata. Food Chem 172:400–406. https://doi.org/10.1016/j.foodchem.2014.09.083
Harnedy PA, O’Keeffe MB, FitzGerald RJ (2017) Fractionation and identification of antioxidant peptides from an enzymatically hydrolysed Palmaria palmata protein isolate. Food Res Int 100:416–422. https://doi.org/10.1016/j.foodres.2017.07.037
Mendis E, Rajapakse N, Kim SK (2005) Antioxidant properties of a radical-scavenging peptide purified from enzymatically prepared fish skin gelatin hydrolysate. J Agric Food Chem 53:581–587. https://doi.org/10.1021/jf048877v
Echave J, Otero P, Garcia-Oliveira P et al (2022) Seaweed-derived proteins and peptides: promising marine bioactives. Antioxidants 11:1–26. https://doi.org/10.3390/antiox11010176
Peighambardoust SH, Karami Z, Pateiro M, Lorenzo JM (2021) A review on health-promoting, biological, and functional aspects of bioactive peptides in food applications. Biomolecules 11:1–21. https://doi.org/10.3390/biom11050631
Cermeño M, Stack J, Tobin PR et al (2019) Peptide identification from a: Porphyra dioica protein hydrolysate with antioxidant, angiotensin converting enzyme and dipeptidyl peptidase IV inhibitory activities. Food Funct 10:3421–3429. https://doi.org/10.1039/c9fo00680j
Fan X, Lv H, Wang L et al (2018) Isoorientin ameliorates APAP-induced hepatotoxicity via activation Nrf2 antioxidative pathway: the involvement of AMPK/Akt/GSK3β. Front Pharmacol 9:1334. https://doi.org/10.3389/fphar.2018.01334
Choi Hee Y, Kim E-Y, Mikami K, Nam Jeong T (2015) Chemoprotective effects of a recombinant protein from Pyropia yezoensis and synthetic peptide against acetaminophen-induced Chang liver cell death. Int J Mol Med 36:369–376. https://doi.org/10.3892/ijmm.2015.2253
Wallace JL (2004) Acetaminophen hepatotoxicity: no to the rescue. Br J Pharmacol 143:1–2. https://doi.org/10.1038/sj.bjp.0705781
Balaji Raghavendra Rao H, Sathivel A, Devaki T (2004) Antihepatotoxic nature of Ulva reticulata (Chlorophyceae) on acetaminophen-induced hepatoxicity in experimental rats. J Med Food 7:495–497. https://doi.org/10.1089/jmf.2004.7.495
Xie Y, McGill MR, Dorko K et al (2014) Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol 279:266–274. https://doi.org/10.1016/j.taap.2014.05.010
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF- 2020R1F1A1074614).
Funding
The authors have no relevant financial or non-financial interests to disclose.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ulagesan, S., Eom, T., Nam, TJ. et al. Antioxidant and chemoprotective peptides from simulated gastrointestinal digested (SGID) protein hydrolysate of Pyropia yezoensis against acetaminophen-induced HepG2 cells. Bioprocess Biosyst Eng 45, 1645–1660 (2022). https://doi.org/10.1007/s00449-022-02770-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-022-02770-4