
Abstract
Climate change and deforestation are causing rainforests to become increasingly fragmented, placing them at heightened risk of biodiversity loss. Invertebrates constitute the greatest proportion of this biodiversity, yet we lack basic knowledge of their population structure and ecology. There is a compelling need to develop our understanding of the population dynamics of a wide range of rainforest invertebrates so that we can begin to understand how rainforest fragments are connected, and how they will cope with future habitat fragmentation and climate change. Blowflies are an ideal candidate for such research because they are widespread, abundant, and can be easily collected within rainforests. We genotyped 188 blowflies (Chrysomya latifrons) from 15 isolated rainforests and found high levels of gene flow, a lack of genetic structure between rainforests, and low genetic diversity – suggesting the presence of a single large genetically depauperate population. This highlights that: (1) the blowfly Ch. latifrons inhabits a ~ 1000 km stretch of Australian rainforests, where it plays an important role as a nutrient recycler; (2) strongly dispersing flies can migrate between and connect isolated rainforests, likely carrying pollen, parasites, phoronts, and pathogens along with them; and (3) widely dispersing and abundant insects can nevertheless be genetically depauperate. There is an urgent need to better understand the relationships between habitat fragmentation, genetic diversity, and adaptive potential–especially for poorly dispersing rainforest-restricted insects, as many of these may be particularly fragmented and at highest risk of local extinction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Due to climate change and deforestation, rainforests are becoming increasingly fragmented islands of endemic biodiversity (Bowman 2000; Lens et al. 2002 Laurance et al. 2017) that are highly susceptible to biodiversity loss (Williams et al. 2003; Mariani et al. 2019; Nolan et al. 2020). To make informed conservation decisions for rainforests and their inhabitants, we must understand how these habitats are fragmented, and how this affects the genetic patterns and ecological processes of their flora and fauna.
Species that inhabit fragmented rainforests can vary significantly in their extent of population genetic structure and diversity – spanning a continuum from well-connected to fragmented populations and low to high genetic diversity (Leung et al. 1994; Brown et al. 2004; Milá et al. 2009; Brito and Arias 2010; Woltmann et al. 2012; Sadanandan and Rheindt 2015). These unique genetic responses to fragmentation result from species-specific physiologies, dispersal capacities, resource requirements, and habitat continua, as well as the landscape structure of the fragments they inhabit (i.e., the presence and number of corridors between adjacent fragments) (Leung et al. 1994; Callens et al. 2011; Woltmann et al. 2012). The consequences of these differences are twofold: (1) certain species will suffer greater genetic and ecological consequences from habitat fragmentation; while (2) other species will play greater roles in maintaining connectivity between fragments. Understanding these species-specific responses is central to quantifying and mitigating further habitat fragmentation under a changing climate.
While attention has focused on many vertebrate animals experiencing anthropogenic threats (Shapcott 2000; Shoo et al. 2005; Mac Nally et al. 2009; Moritz et al. 2005; Martínez-Ramos et al. 2016), invertebrates have been largely neglected (Ellwood and Foster 2004; Snaddon 2008; Wardhaugh et al. 2012; Taylor et al. 2018; New 2018). Of all animals, invertebrates are the largest contributors to the function and biodiversity of rainforest ecosystems (Ewers et al. 2015; Griffiths et al. 2017) and perform crucial roles in maintaining habitat connectivity by promoting gene flow in their associated plants and parasites (Jabis et al. 2011; DiBlasi et al. 2018). Despite this, we have a very limited understanding of the population structure of most rainforest invertebrates (Radespiel and Bruford 2014), including whether there is connectivity/gene flow among geographically disconnected fragments, and which species and populations require conservation priority. There is an urgent need to remedy this, as studies around the world are beginning to identify significant changes in invertebrate populations, with habitat fragmentation and climate change as key drivers (Cardoso et al. 2020; Samways et al. 2020; Legge et al. 2021; Marsh et al. 2021).
Australian rainforests are naturally fragmented due to the unique geological and climatic history of the continent (Bowman 2000). However, many rainforests have also been further fragmented by human processes since European colonisation (Bowman 2000) as well as due to increasing rates of wildfires (Legge et al. 2021; Trouvé et al. 2021). Importantly, these highly fragmented rainforests hold a substantial proportion of the continent’s total invertebrate biodiversity (Kitching et al. 1993; Yeates et al. 2003; Stork and Grimbacher 2006). As such, there is a need to understand how invertebrates are distributed throughout these rainforests, determine whether there is gene flow between populations, and identify which species and populations are less likely to cope with sudden changes to climate or habitat structure (Kelly 2019; Razgour et al. 2019) and which contribute most to maintaining connectivity between habitat fragments. However, given the overwhelming diversity of rainforest invertebrates, it is not currently feasible to comprehensively assess the genetic responses of every species to habitat fragmentation. The simplest solution is to begin studying species that inhabit fragmented rainforests but are broadly distributed throughout them, can easily disperse between them, and can be reliably collected. Such species should show generally high levels of genetic connectivity between habitat fragments, providing a baseline understanding of how rainforest fragments are connected, and which are most isolated, to inform how they might cope with further fragmentation.
Blowflies (Diptera: Calliphoridae) are well-suited to this approach because they are widespread throughout rainforests, extremely abundant, and easy to capture in the wild (Norris 1959; Badenhorst and Villet 2018; Butterworth et al. 2020). Blowflies are also highly vagile (Norris 1959; Tsuda et al. 2009) and many species exhibit wide dietary breadths – opportunistically feeding on a range of resources, including vertebrate and invertebrate carrion, dung, decaying plant matter, and pollen (Dear 1985; Brodie et al. 2015). As such, blowflies are likely some of the most capable invertebrate dispersers and can be expected to play important roles in maintaining connectivity between even the most highly isolated habitat fragments.
Here, we perform extensive field collections throughout rainforest fragments in southeast Australia and use genotype-by-sequencing (GBS) through the DarTseq™ platform to obtain genetic information (single nucleotide polymorphisms; ‘SNPs’), targeting a species that is endemic to the region – Chrysomya latifrons Malloch 1927 (Fig. 1). This highly mobile species is frequently collected in fragmented rainforests across a large (~ 1000 km) geographic range. We expect that Ch. latifrons should show high levels of gene flow and limited genetic differentiation between adjacent rainforests. As we expect to observe only broad patterns of isolation by distance, any rainforest fragments inhabited by Ch. latifrons that do not meet these patterns may reflect high levels of habitat isolation.

A The blowfly Chrysomya latifrons which are endemic to southeast Australia and abundant in rainforests; and B the typical rainforest habitat where they can be found (pictured: Washpool National Park, NSW, Australia). Photographs were taken by NB
Methods
Study species
Ch. latifrons has generally been considered a rainforest specialist that is restricted to New South Wales (NSW), Australia (Malloch 1927; Wallman 2002) and has been commonly recorded throughout rainforests ranging from Wollongong to Coffs Harbour (Butterworth et al. 2020). However, it has also been collected in rainforest-adjacent dry eucalypt forests (Dawson et al. 2021), as well as in urban green spaces around the Sydney region (Kavazos and Wallman 2012). As such, Ch. latifrons appear to be capable of tolerating a wide range of habitats but most commonly encountered in rainforests.
Specimen collecting
To create a bait that was attractive to Ch. latifrons, 500 g of raw kangaroo mince was left outdoors for 72 h to attract Calliphora blowflies. Once Calliphora larvae had fed on the meat and reached the third instar, we tightly sealed the container for 24 h. The subsequent anaerobic decomposition of Calliphora maggots (in combination with the decomposing kangaroo mince) results in the production of specific volatiles that are highly attractive to all Australian Chrysomya species (except Chrysomya flavifrons Aldrich 1925). This bait presumably exploits the preference of Chrysomya species for cues associated with carrion that is in the mid-stage of decomposition, as most Chrysomya are secondary colonisers (Dawson et al. 2021).
Baits were used to collect a total of 188 Ch. latifrons adults with sweep nets at rainforest sites between Washpool, NSW and Maxwells Rainforest, NSW (for all rainforest sampling locations refer to Table 1, Fig. 2). Sampling trips were conducted between November 2020 and February 2021, and specimens were only collected at each site during a single round of sampling (between 2 to 6 h per site). The collection location at each rainforest was chosen at random and was not expected to bias signatures of genetic structure, as blowflies are strong dispersers and are finely tuned to the detection of carrion (Norris 1959), so any blowflies caught on the bait will broadly represent that rainforest’s genetic diversity. Flies were euthanised with ethyl acetate vapour within eight hours of capture, taxonomically identified following the key of Wallman (2002), placed into 2.5 mL plastic tubes containing 90% ethanol, and stored at − 4 °C in the laboratory for up to eight weeks.

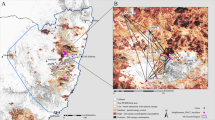
Spatial structure of rainforests (represented by dark green) throughout New South Wales, Australia. Data sourced from the Australian State of the Forests 2018 report (https://www.agriculture.gov.au/abares/forestsaustralia/sofr/sofr-2018). Rainforest habitat was defined as “Forest dominated by broad-leaved tree species, typically in wet or sheltered environments and with a closed canopy. Can include areas with non-rainforest species as emergents (trees emerging above the canopy), but where rainforest species dominate the character of the site”. Spatial fragmentation of rainforests at the local scale (10 km) is also represented for three populations (WA, OU, MA) in the right panels
Rainforest characteristics
To map the spatial distribution of rainforest fragments and to estimate levels of rainforest fragmentation, we downloaded the dataset from the Australian State of the Forests 2018 report (https://www.agriculture.gov.au/abares/forestsaustralia/sofr/sofr-2018), which characterises rainforests as “Forest dominated by broad-leaved tree species, typically in wet or sheltered environments and with a closed canopy. Can include areas with non-rainforest species as emergents (trees emerging above the canopy), but where rainforest species dominate the character of the site”. We then used QGIS version 3.28.1 (http://www.qgis.org/) to filter this dataset on the rainforest, exported this data from raster to polygons for mapping, clipped to the extents of NSW, and created a 10 km and 60 km buffer around each site. We chose both scales as they represent the range of observed dispersal capacities of Chrysomya blowflies (Norris 1959; Braack and Retief 1986). By incorporating both spatial scales, we can, therefore, see how the extent of patch connectivity and fragmentation change within the spatial extent that is relevant to the focal organism – where truly ‘isolated’ patches should show a low number of fragments and total area at both spatial scales. We intersected both the 10 km and 60 km buffers with the rainforest dataset to return the total number of rainforest patches and dissolved these resulting features to get estimates of the total rainforest area. Lastly, we divided the rainforest area by count to calculate the average size of patches based on the 10 km and 60 km buffers. Together, these characteristics provide proxies of rainforest fragmentation surrounding each collection locality – where fewer patches, less total rainforest area, and lower average patch sizes represent higher levels of fragmentation.
DArTSeq™ assay
The head of each individual fly (3–18 from each population) was dissected from the body, placed in a single well of a 96-well plate with 70% ethanol, and supplied to Diversity Arrays Pty Ltd (Canberra, Australia) for a high-density DArTSeq™ assay (~ 2.5 million sequences/sample used in marker calling). The DArTSeq™ extraction and sequencing methods are detailed in Kilian et al. (2012) and Georges et al. (2018). In brief, restriction enzyme digestion is utilised to digest genomic DNA into fragments which are then sequenced using an Illumina short-read platform. Short-read sequences are then bioinformatically processed to produce a series of SNPs.
To ensure appropriate DNA fragments were used for subsequent sequencing, restriction enzyme digestion was optimised for Ch. latifrons using multiple restriction enzyme combinations and eight specimen replicates. Following sequencing of these test specimens, the optimal restriction enzyme pair was identified as PstI-HpaII, based on the fraction of the genome represented, while controlling for average read depth and the number of polymorphic loci. This restriction enzyme combination was used for all subsequent digestions. Following digestion, all sequence fragment libraries were ligated with Illumina sequencing adaptors and sequenced on an Illumina HiSeq2000 platform.
Short-read sequences were processed using the DarTseq™ bioinformatic pipeline (Georges et al. 2018), which performs filtering and variant calling, to generate final genotypes. While some parts of the sequencing and analysis protocol cannot be provided, the use of the DArTSeq platform for studies of genetic diversity and structure is widespread (Popa‑Báez et al. 2020; Hoffman et al. 2021; Jaya et al. 2022) and is reproducible because subsequent use of this proprietary service will yield the same results.
SNP filtering
The DArTseq™ dataset contained a total of 21006 SNPs across 187 individual flies (Supplementary material 1), as a single specimen from Washpool National Park was removed from the dataset due to low quality reads. The data were then filtered with the ‘dartR’ package version 1.9.9.1 (Gruber et al. 2018) in R version 3.6.1 (R Core Team 2019). It is generally recommended that a range of data filtering thresholds are applied and tested for the same dataset (Wright et al. 2019; Schmidt et al. 2021), particularly because optimal metrics and thresholds will differ between analyses (Linck and Battey 2019; Hoffmann et al. 2021). As such, we first filtered the DArTseq™ dataset by reproducibility (proportion of technical replicate assay pairs for which the marker score was consistent) at a threshold of 0.98, then by call rate (proportion of samples for which the genotype call was not missing) at a threshold of 0.95, and finally by minor allele count (MAC less than the threshold are removed) at a threshold of 3. This resulted in a filtered dataset of 187 individual genotypes, 3910 SNPs, and 1.22% missing data. To compare the impact of MAC vs minor allele frequency (MAF) filtering, we next filtered the original dataset using the same parameters above but with a MAF threshold of 0.02 instead of a MAC of 3. This resulted in a final dataset of 187 individual genotypes, 2693 SNPs, and 1.25% missing data, which was used for all downstream analyses. As has been recommended for analyses of population structure (Linck and Battey 2019; Hoffmann et al. 2021), we used the MAC-filtered DArTseq™ dataset as a comparison for later admixture analyses, and the MAF-filtered dataset for all other downstream analyses (see results).
To consider the effect of assembly parameters on alignments and downstream analyses (e.g., Linck and Battey 2019), we also processed the raw fastq files from DArT-Seq™ samples with IPYRAD v. 0.7.28 (Eaton and Overcast 2020), to filter and remove low-quality data, identify homology among reads through de novo assembly, make SNP calls, and create a variant call format (VCF) file of variant sites (https://ipyrad.readthedocs.io/en/master/). Default settings were used in IPYRAD, with the exception of the following: strict filtering of adapters was applied (filter_adapters = 2; default 0—no filtering); and the final minimum length after filtering was set to 60 (filter_min_trim_len = 60; default 35). The IPYRAD dataset contained a total of 48,912 SNPs across 187 individual flies. The data were then filtered in VCFTOOLS v. 0.1.13 (Danecek et al. 2011) using –missing-indv, –max-missing-count, and –maf filters to remove any individuals with more than 50% missing data, any sites with more than 20% missing genotypes, and any sites with a MAF of less than 5%, respectively. This resulted in a final dataset of 7862 SNPs.
Genetic diversity
We used R for all analyses of genetic diversity, and unless otherwise stated, all analyses were conducted using the DArTseq™ genotype dataset filtered for a MAF of 0.02. We first applied the ‘basic.stats’ function of the ‘hierfstat’ package version 0.5–10 (Goudet et al. 2015) to calculate average observed heterozygosity (HO), expected heterozygosity (HE), and inbreeding coefficients (FIS). We also used the ‘betas’ function from ‘hierfstat’ to calculate population-specific FST values (Weir and Goudet 2017).
Population structure
Using R, we first assessed population structure by AMOVA using the function ‘poppr.amova’ with the ‘ade4’ implementation from the ‘poppr’ package version 2.9.3 (Kamvar et al. 2014). To test whether populations were significantly different, we used a randomisation test on the AMOVA output with 1000 permutations (Excoffier et al. 1992) using the function ‘randtest’ from the package ‘ade4’ version 1.7–18 (Thioulouse et al. 2018). We then conducted pairwise comparisons of FST values between populations using the ‘gl.fst.pop’ function from the ‘dartR’ package with 10,000 bootstrap replicates.
Genetic distances between individuals were examined using Nei’s distances, and a dendrogram with 1000 bootstrap replicates was created with the ‘aboot’ function of the ‘poppr’ package, and the ‘ggtree’ function of the package ‘ggtree’ (Yu 2020). We then used the ‘glPca’ function from the ‘adegenet’ package version 2.1.5 (Jombart 2008) to determine whether genetic differences between individuals (as represented by principal components) were structured according to their populations.
To test for isolation by distance, we performed a Mantel test using the function ‘gl.ibd’ from the ‘dartR’ package in R. This compared linearised genetic distances between populations (calculated using ‘StAMPP’ version 1.6.3; Pembleton et al. 2013) against Euclidean geographical distances (calculated using ‘vegan’ version 2.5–7; Oksanen et al. 2013). We followed this with a mantel correlogram analysis using the ‘vegan’ function ‘mantel.correlog’.
To calculate individual blowfly admixture coefficients, the MAF-filtered SNP data were converted into the STRUCTURE format (‘.str’) using the ‘gl2faststructure’ function from the ‘dartR’ package, then into the ‘.geno’ format using the ‘struct2geno’ function of the ‘LEA’ package version 3.1.4 (Frichot and Francois 2015). We then ran sparse non-negative matrix factorisation on these data with the ‘sNMF’ function from ‘LEA’. We analysed K values of 1 to 10, with 100 replications for each K value, and used the cross-entropy criterion to determine the value of K that best explained the results. To assess whether filtering had a major impact on signatures of population structure, we ran this analysis a second time on the DArTseq™ dataset after filtering for a MAC of 3, as has been recommended for analyses of population structure (see above).
To verify these results across our different bioinformatic pipelines, we also analysed the IPYRAD-filtered dataset using sNMF (with the same parameters as above) and tested the IPYRAD-filtered dataset with the program STRUCTURE v.2.3.4 (https://web.stanford.edu/group/pritchardlab/structure.html) across a K value range of 1 to 10 with 10 replications per K. An initial analysis was completed to determine the number of Markov Chain Monte Carlo (MCMC) iterations required to achieve stationarity in FST and log(α) summary statistics for each K value. The optimal configuration was determined to be 100,000 MCMC iterations, with 5000 discarded as burn in and these parameters were used for all subsequent analyses. Following STRUCTURE analysis, the best K value was selected using the Evanno method (Evanno et al. 2005) implemented within the Structure Harvester online interface (available at: http://taylor0.biology.ucla.edu/structureHarvester/). To create plots, results for each K value were permuted across all replicates using CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007) and then plotted using Toyplot 0.18.0 (https://github.com/sandialabs/toyplot).
Gene flow
Finally, to calculate relative migration rates among populations, we used the divMigrate implementation (Sundqvist et al. 2016) of the ‘diveRsity’ package version 1.9.90 (Keenan et al. 2013) in R. The directional migration calculations implemented by divMigrate require that populations are predetermined (Sundqvist et al. 2016), however, we were unable to make any population assignments based on the sNMF or STRUCTURE analyses. As such, we performed two separate analyses: the first with all 15 geographic populations, and the second with individuals only from the northern-most population (WA), the central population (KE), and the southern-most population (MA) – on the assumption that these are the most geographically isolated and should show the lowest levels of directional gene-flow. For both analyses, we estimated 50 bootstrap iterations, set the filter threshold to 0.2, and chose the effective number of migrants (Nm) to infer levels of connectivity between populations.
Results
Rainforest fragmentation is extensive at both local scales of 10 km and broader scales of 60 km, with often hundreds of small rainforest patches spread throughout drier sclerophyll forests. Fragmentation is particularly pronounced at lower latitudes (Fig. 2; Table 2). We see that Wyrrabalong National Park (WY) which is composed of one major rainforest patch and appears relatively isolated at a scale of 10 km is in fact relatively well connected at a scale of 60 km (which is within the dispersal kernel of Ch. latifrons). Alternatively, Maxwell’s Rainforest (MA) which appears relatively well connected at a scale of 10 km is in fact quite isolated at a scale of 60 km – and would thus be expected to have the lowest level of broad connectivity to other patches. Nevertheless, at most sites, Ch. latifrons were abundant at the time of collection, ranging from a constant number of five to 20 individuals on the carrion bait. However, at Ulidarra National Park (UL) we only observed three Ch. latifrons as the community was dominated by Ch. semimetallica – suggesting this may be close to the range edge of Ch. latifrons. At Wyrrabalong National Park (WY) we only observed five flies throughout four hours of collecting (Table 2).
Genetic diversity
Unless otherwise stated, all results relate to the DArTseq™ dataset filtered for a MAF of 0.02. Across all SNP loci, 75% of individuals were homozygous for the reference allele, 7% were homozygous for the alternate allele, and 18% were heterozygous – indicating relatively low genetic diversity across all populations. Low heterozygosity was also observed within each population, with population-specific HO values all < 0.180, and all lower than the average expected heterozygosity (HE) (Table 1). Further to this, more than 30% of loci had an average HO of ≤ 0.10, and only 20% of loci had an average HO > 0.30 (Supplementary material 2: Fig. 1). Despite HO being less than HE in all populations, no strong signatures of inbreeding were detected, with low FIS values in each population (always < 0.150; Table 1).
Population structure
Population-specific FST values were low (ranging from 0.000 to 0.032), suggesting minimal genetic differentiation between populations. Likewise, pairwise comparisons of FST values between populations were all < 0.009, and there were no significant differences detectable between geographic populations (Table 3). AMOVA indicated that most of the genetic variation was observed within individuals (83.5%) followed by between individuals (16.5%), and both were found to be significant factors contributing to the overall population structure (P < 0.01) (Table 4). However, genetic variation between populations was not detected (0.00%), consistent with the FST results.
There was also no clear differentiation between individuals from different populations based on Nei’s genetic distances. In fact, individuals from populations separated by > 900 km often showed greater genetic similarity to each other than to individuals that were collected from the same geographic population (Fig. 3). Consistent with this, both the Mantel test of genetic versus geographic distance and the Mantel correlogram yielded no significant correlations (Mantel’s r = − 0.053, P = 0.685) (Supplementary material 2: Fig. 2a, b). Likewise, in the principal component analysis (Fig. 4, Supplementary material 2: Fig. 3), the first two principal components explained only 0.97% and 0.95% of the total variation, and there was no clear separation of populations based on individual principal component scores (Fig. 3).

A dendrogram based on Nei’s genetic distances for individual Chrysomya latifrons (Diptera: Calliphoridae) from 15 rainforest populations. Populations are coloured according to the provided key in order from north (dark purple) to south (yellow). Population abbreviations are provided in Table 1

Principal component analysis of the filtered dataset of 2693 SNP loci from 15 populations of Chrysomya latifrons (Diptera: Calliphoridae). Populations are coloured according to the provided key in order from north (dark purple) to the south (yellow). Population abbreviations are provided in Table 1
The sNMF analyses of all three datasets (MAF = 0.02, MAC = 3, and IPYRAD) all identified the lowest cross-entropy for a K value of one (Supplementary material 2: Fig. 4), and all showed similar admixture proportions (Supplementary material 2: Fig. 5) suggesting that all geographic populations share a common ancestry. To investigate admixture in more detail, we explored the results from the MAF = 0.02 filtered dataset for K-values from 1 to 10. For all tested K-values > 1, we found that admixture was evenly spread throughout geographic locations – supporting the notion that all populations share a common ancestry and that none were substantially genetically differentiated. To visualise these admixture proportions, we present the results for K = 2 in Fig. 5.

The mean admixture proportions of populations of Chrysomya latifrons (Diptera: Calliphoridae) that were sampled in the present study. The admixture proportions plotted on the map represent population averages. The bar plots presented on the right reflect individual admixture proportions, sorted by population, where each bar represents a single individual. Full population names are provided in Table 1. All analyses and plotting can be replicated by following the online tutorial provided by Tom Jenkins (https://github.com/Tom-Jenkins/admixture_pie_chart_map_tutorial)
Likewise, the STRUCTURE analysis based on the IPYRAD dataset (Fig. 6a) was concordant with the sNMF analyses and suggested no clear genetic differentiation between populations at K = 3 (the optimal K value supported by the Evanno method).

Geographic distribution of genetic structure and gene flow in Chrysomya latifrons. Full population names are provided in Table 1. a STRUCTURE plot of genetic assignment (K = 3) for 187 individuals. Populations are sorted from top to bottom geographically (north to south). b A relative migration network. Letters represent sampling sites, arrows mark the direction of gene flow, and numbers represent the level of migrant exchange between locations (i.e., effective number of migrants Nm) (Sundqvist et al. 2016). Results of the same analysis with all 15 geographic populations are presented in Supplementary material 2: Table 1
Gene flow
Gene flow was generally high among all 15 populations (Supplementary material 2: Table 1), with numbers of effective migrants (Nm) > 0.5 with the exception of the WY and UL populations (which both had low sample sizes of N ≤ 5), and with no significant asymmetries in migration rates between population pairs. When considering only the three most isolated populations (WA, KE, and MA), gene flow was again determined to be high in all cases (Nm ≥ 0.74) with no significant asymmetries in migration rates between population pairs (Fig. 6b).
Discussion
We assessed genetic connectivity among fragmented rainforests in the endemic Australian blowfly Ch. latifrons. Based on the ubiquity, widespread distribution, and high dispersal ability of Ch. latifrons, we expected to find limited genetic differences between adjacent rainforests and only a broad pattern of isolation by distance. However, we found a complete lack of genetic structure between populations – indicative of a single large, panmictic population despite the ~ 1000 km range distribution span. Broadly, this highlights that strongly dispersing insects, such as blowflies, may have the capacity to disperse between and connect isolated rainforest fragments.
Distribution and biology
We found Ch. latifrons in high abundances (at the time of collection) from Washpool National Park in northern NSW to Maxwells Rainforest on the southern border with Victoria. This is the first comprehensive information on the distribution of Ch. latifrons, which inhabits a large latitudinal transect of > 1000 km of eastern Australian rainforests and rainforest-adjacent sclerophyll forests. It is almost certain that populations also extend into the rainforests of northern Victoria, where there is suitable habitat available. Remarkably however, Ch. latifrons was completely absent near the Queensland border, where its closest genetic relative, Ch. semimetallica, occurs (Butterworth et al. 2020). These two species are rarely collected at the same location and likely compete with each other in a thin hybrid zone between Coffs Harbour in NSW and Brisbane in Queensland – providing an interesting model system for future work focused on understanding the eco-evolutionary dynamics of allopatry.
It was clear from our sampling that Ch. latifrons inhabits every major rainforest in south-eastern Australia, often greatly outnumbering other carrion-breeding flies on a given resource during the days we sampled – it was not uncommon to see 30–40 individuals around larger resources in the mid-stages of decomposition. Importantly, however, our observations of abundance are based only on one-time point. Although consistent with high numbers observed in previous studies (Kavazos and Wallman 2012; Butterworth et al. 2020; Dawson et al. 2021), there is likely to be significant year-to-year variation in abundances as well as temporal variability throughout the day. Without more extensive and repeated sampling, we are unable to make any general conclusions on the abundance of Ch. latifrons. In fact, this is the case for most rainforest invertebrates, particularly decomposers. There is still much to learn about seasonal and temporal drivers of the distribution and abundance of invertebrates in Australia; such knowledge will be instrumental to understanding the functioning of Australian ecosystems.
Considering the widespread distribution of Ch. latifrons and its common occurrence in rainforest carrion, it is likely playing an important role in rainforest ecosystems as a primary decomposer, but its biology is poorly understood. It is likely to be a carrion-breeding species, infesting vertebrate carrion in rainforests and adjacent habitats. This is supported by its attraction to carrion baits, oviposition on carrion, and the similarity of its larvae to other closely related carrion-breeding generalist species (Szpila and Wallman 2016). The widespread distribution and ability of Ch. latifrons to easily disperse long distances between habitats (see below) suggests they are either generalist consumers of a wide range of animal carrion types, or specialist consumers that utilise types of carrion that are abundant throughout rainforests.
Genetic diversity and population structure
Values of observed heterozygosity < 0.30 are considered to represent low genetic diversity across a wide range of animals (Andere et al. 2008, 2020; Robertson et al. 2018; Kleinhans and Willows-Munro 2019; Melo-Carrillo et al. 2020). For all the populations sampled in the present study, the mean observed heterozygosity was < 0.18. In fact, more than 30% of loci had an observed heterozygosity of ≤ 0.10, and observed heterozygosity was consistently lower than expected heterozygosity. These values of observed heterozygosity are particularly low when compared to many other strongly-dispersing fly species, where average levels of observed heterozygosity in microsatellite markers between 0.3 and 0.7 have been recorded (Wilke et al. 2017; Qin et al. 2018; Bateta et al. 2020; Deschepper et al. 2021). Low levels of heterozygosity seem unlikely to be due solely to inbreeding, as signatures of inbreeding were not strong based on FIS. Rather, they may simply be the result of significant historical bottlenecks (Menken 1987; Brouwer et al. 2007) or selection against heterozygotes (underdominance) (Láruson and Reed 2016). Broadly, this suggests a widespread lack of genetic diversity for Ch. latifrons, despite these flies being readily abundant during sampling, and spread over such a wide geographic range. In addition, population-specific FST values indicate that population structure in Ch. latifrons is extremely low or absent, and this was reinforced by the consistent results of AMOVA, Nei’s genetic distances, principal component analysis, isolation-by-distance, admixture, and STRUCTURE analyses. Taken together, the diversity and differentiation results suggest that Ch. latifrons exist within east Australian rainforests as one large panmictic and genetically depauperate population.
The low genetic diversity and lack of population structure observed in Ch. latifrons highlights that widely dispersing and abundant insects can nevertheless be genetically depauperate. Although it is expected that species competing on ephemeral resources should maintain high genetic diversity (Dytham and Shorrocks 1992), hypotheses regarding resource dynamics and population structure have rarely been tested in invertebrate populations. The unpredictable nature of the larval resources (which can also be adult mating sites) may lead to sweepstakes reproduction where population sizes are large due to high fecundity, but only a small number of individuals are able to successfully reproduce. Similar patterns of limited genetic diversity due to sweepstakes reproduction have been shown in other insects (Ruiz-Carbavo 2022).
Genetic diversity is expected to be closely linked to the adaptive capacity of populations (Hoffmann et al. 2017), so even though Ch. latifrons is widespread, it may be threatened by the drastic climatic and environmental changes (e.g., increasing fires and temperature; Legge et al. 2021) that are occurring along its habitat range. Particularly if these exert significant pressure on its limited pool of genetic diversity. A direct link between low genetic diversity and low adaptive potential has not been confirmed for most terrestrial invertebrates (Kellerman and van Heerwaarden 2019). However, the Australian fly species Drosophila birchii inhabits a similar stretch of rainforest fragments and, despite being widespread, has been shown to have limited additive genetic variance for desiccation resistance (Kellerman et al. 2006). Future studies should aim to characterise the adaptive capacity of Ch. latifrons (and a wider range of rainforest invertebrates), particularly in the context of tolerance to climate change – as rainforests continue to experience its increasingly severe and frequent effects (Jiang et al. 2019; Boulton et al. 2022). Such research should not only focus on rare or endangered species but also on common and widely distributed species that may play important roles in maintaining habitat connectivity between fragments.
Potential explanations for lack of diversity and population structure
The low population structure we observed in Ch. latifrons is unlikely to solely be a result of their strong dispersal ability, as there is ample evidence that flying insects with strong dispersal capacities often still show high levels of population structure (e.g., Bateta et al. 2020; Bluher et al. 2020). One possible explanation may relate to migration patterns. Chrysomya species tend to thrive in warmer temperatures (Byrd and Butler 1997; Sontigun et al. 2018), so it is plausible that a major source population is maintained in the warmer northern rainforests during winter, from which numerous individuals may migrate towards southern rainforests over spring and summer.
This phenomenon is suggested to occur in Australian Chrysomya rufifacies (Norris 1959) as well as American populations of the blowfly Cochliomyia hominivorax (Eddy and Bushland 1956). Blowflies have been shown to disperse as far as 6 km within 24 h (Norris 1959), while some Chrysomya species can travel up to 65 km (Braack and Retief 1986), and individual stable flies (Stomoxys calcitrans) can migrate as far as 225 km (Hogsette and Ruff 1985). Thus, it is possible that long-distance migration from a small winter source population explains the lack of population structure of Ch. latifrons. In fact, many Australian insects are known to migrate great distances and change distributions between seasons. For example, Mythimna and Helicoverpa moths (Lepidoptera) both make long south-easterly migrations in the warmer spring and summer months (Drake and Gatehouse 1995; Satterfield et al. 2020).
Strongly dispersing species with long-distance seasonal migration patterns generally tend to show low population structure over broad geographic ranges (Endersby et al. 2007; Pfeiler and Markow 2017; Wang et al. 2021), further supporting the possibility that large numbers of Ch. latifrons undertake long migrations during the spring and summer. There are numerous rainforest pockets and corridors that could facilitate such migrations, particularly along the mountainous Great Dividing Range, which extends from Queensland to Victoria. However, our understanding of insect migration is still limited (Chapman et al. 2015; Pfeiler and Markow 2017; Satterfield et al. 2020), particularly as widespread distributions of insect populations in the summer months may mask the risk of local extinction of winter populations. This could occur in Ch. latifrons, particularly if a major source population is maintained in only a few northern NSW rainforests during the winter.
The spatial structure of carrion resources may also play a role, as these are often used as mating sites by blowflies (Butterworth et al. 2018, 2019). Carrion patches may thus act as ecological islands (Barton et al. 2013) between fragmented rainforests, facilitating genetic connectivity between patches at both local and broader scales. Physiological aspects may also play a role in driving population structure in Ch. latifrons. Some Chrysomya species can cope with temperatures as low as 11 °C (Cammack and Nelder 2010), and Ch. latifrons can occur around the greater Sydney region during the winter (Kavazos and Wallman 2012; Dawson et al. 2021). Individuals may diapause in southern regions throughout the winter (Saunders and Hayward 1998; Vinogradova and Reznik 2013), although there is no current evidence that Ch. latifrons has this capacity. Alternatively, the temperature and climatic differences between northern and southern rainforests during winter may not be drastic enough to prevent Ch. latifrons from inhabiting southern rainforests throughout the entire year. In this case, panmixia may be the result of more continuous patterns of breeding between rainforests along eastern Australia. A lack of genetic diversity and population structure has also been shown along a north–south transect of Queensland fruit fly populations (Popa‑Báez et al. 2020), supporting the idea that temperature variation along south-eastern Australia may not markedly constrain fly movement.
Finally, the lack of population structure in Ch. latifrons may not reflect the current population dynamics of the species. In the case of recent fragmentation (due to deforestation or wildfires) and large enough population sizes, there may not have been sufficient time for a resulting lack of gene flow to be reflected in patterns of population structure and diversity (i.e., Lloyd et al. 2013; Su et al. 2018; Delnevo et al. 2021). However, this seems unlikely, as Ch. latifrons can be found in habitats adjacent to rainforests (i.e., dry schlerophyll forests and urban areas) and almost certainly has the capacity to disperse through rainforest-adjacent habitats. Importantly, the lack of population structure we observed does not necessarily imply that there are no signatures of local adaptation in Ch. latifrons. Such questions could be addressed by measuring phenotypic differences between populations or with other sequencing approaches (e.g., genome skimming, ultra-conserved elements, exome, transcriptome) to investigate fine-scale signatures of local adaptation.
Implications for rainforest connectivity
Our results provide important insights into broad patterns of rainforest connectivity in Australia – highlighting that rainforest inhabitants with strong dispersal capacities (such as blowflies) can likely move between, and connect highly fragmented habitats. Such widespread dispersal between rainforests is unlikely to be representative of all strong dispersers, as it depends largely on species-specific niche breadths and resource distributions that may constrain certain species to specific rainforest patches (e.g., Woltmann et al. 2012).
However, the ubiquitous distribution and movement of Ch. latifrons between isolated rainforests makes it a likely vector of dispersal for pollen, pathogens, parasites, and phoronts – forming an interconnected ecological network throughout southeast Australian rainforests. As such, we may also expect to see high degrees of genetic connectivity between the rainforest taxa that are associated with Ch. latifrons. Such correlated patterns of dispersal between fragmented habitats have been shown for rainforest trees and the African honeybees that pollinate them (Dick et al. 2003). Future work should, therefore, target the ecology of Ch. latifrons, particularly to understand their broad contributions to rainforest ecosystems and their adaptive potential in a changing climate.
Conclusions
The future capacity of species to disperse between rainforests is likely to be hampered by ongoing climate change and deforestation, particularly with the increased likelihood of climatic disturbances (i.e., wildfires: Legge et al. 2021). Within Australia, the long-term effects of the devastating 2019–2020 bush fires on habitat fragmentation and population dynamics are yet to be fully seen, and the data we present for Ch. latifrons is a crucial reference point for long-term studies aimed at understanding these dynamics. There is, however, an urgent need to study the genetic structure and adaptive capacity of more poorly dispersing rainforest insects that are endemic to fragmented rainforests (Kellerman and van Heerwarden 2019). These insects are likely to be highly adapted to their specific habitats and are perhaps at the highest risk of local extinction due to climate change. Potential candidates are millipedes, worms, snails, ticks, spiders, and springtails – all of which are widely dispersed through rainforests (Mesibov 1998; Olson 1994) but lacking information on population genetic structure and adaptive capacity. We strongly encourage researchers to focus on such target species to further understand rainforest population dynamics and conservation priorities.
Data availability
Individual genotype data (SNP 1-row format) and sample metadata are available as supplementary data.
Code availability
Available upon request.
Change history
03 March 2023
Missing Open Access funding information has been added in the Funding Note.
References
Andere AA, Pimsler ML, Tarone AM, Picard CJ (2020) The genomes of a monogenic fly: views of primitive sex chromosomes. Sci Rep 10:15728. https://doi.org/10.1038/s41598-020-72880-0
Badenhorst R, Villet MH (2018) The uses of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae) in forensic entomology. Forensic Sci Res 3:2–15. https://doi.org/10.1080/20961790.2018.1426136
Barton PS, Cunningham SA, Lindenmayer DB, Manning AD (2013) The role of carrion in maintaining biodiversity and ecological processes in terrestrial ecosystems. Oecologia 171:761–772. https://doi.org/10.1007/s00442-012-2460-3
Bateta R, Saarman NP, Okeyo WA, Dion K, Johnson T, Mireji PO, Okoth S, Malele I, Murilla G, Aksoy S, Caccone A (2020) Phylogeography and population structure of the tsetse fly Glossina pallidipes in Kenya and the Serengeti ecosystem. PLOS Neglect Trop D. https://doi.org/10.1371/journal.pntd.0007855
Bluher SE, Miller SE, Sheehan MJ (2020) Fine-scale population structure but limited genetic differentiation in a cooperatively breeding paper wasp. Genome Biol Evol 12:701–714. https://doi.org/10.1093/gbe/evaa070
Boulton CA, Lenton TM, Boers N (2022) Pronounced loss of amazon rainforest resilience since the early 2000s. Nat Clim Chang 12:271–278. https://doi.org/10.1038/s41558-022-01287-8
Bowman DMJS (2000) Australian rainforests: islands of green in a land of fire. Cambridge University Press, United Kingdom
Braack LE, Retief PF (1986) Dispersal, density and habitat preference of the blow-flies Chrysomyia albiceps (Wd.) and Chrysomyia marginalis (Wd.) (Diptera: Calliphoridae). Onderstepoort J Vet Res 53:13–18
Brito RM, Arias MC (2010) Genetic structure of Partamona helleri (Apidae, Meliponini) from neotropical atlantic RAINFOREST. Insect Soc 57:413–419. https://doi.org/10.1007/s00040-010-0098-x
Brodie BS, Smith MA, Lawrence J, Gries G (2015) Effects of floral scent, color and pollen on foraging decisions and oocyte development of common green bottle flies. PLoS ONE. https://doi.org/10.1371/journal.pone.0145055
Brouwer L, Komdeur J, Richardson DS (2007) Heterozygosity–fitness correlations in a bottlenecked island species: a case study on the Seychelles warbler. Mol Ecol 16:3134–3144. https://doi.org/10.1111/j.1365-294X.2007.03370.x
Brown LM, Ramey RR, Tamburini B, Gavin TA (2004) Population structure and mitochondrial DNA variation in sedentary neotropical birds isolated by forest fragmentation. Conserv Genet 5:743–757. https://doi.org/10.1007/s10592-004-1865-x
Butterworth NJ, Byrne PG, Keller PA, Wallman JF (2018) Body odor and sex: do cuticular hydrocarbons facilitate sexual attraction in the small hairy maggot blowfly? J Chem Ecol 44:248–256. https://doi.org/10.1007/s10886-018-0943-3
Butterworth NJ, Byrne PG, Wallman JF (2019) The blow fly waltz: field and laboratory observations of novel and domplex dipteran courtship behavior. J Insect Behav 32:109–119. https://doi.org/10.1007/s10905-019-09720-1
Butterworth NJ, Wallman JF, Drijfhout FP, Johnston NP, Keller PA, Byrne PG (2020) The evolution of sexually dimorphic cuticular hydrocarbons in blowflies (Diptera: Calliphoridae). J Evol Biol 33:1468–1486. https://doi.org/10.1111/jeb.13685
Byrd JH, Butler JF (1997) Effects of temperature on Chrysomya rufifacies (Diptera: Calliphoridae) development. J Med Entomol 34:353–358. https://doi.org/10.1093/jmedent/34.3.353
Callens TOM, Galbusera P, Matthysen E, Durand EY, Githiru M, Huyghe JR, Lens LUC (2011) Genetic signature of population fragmentation varies with mobility in seven bird species of a fragmented Kenyan cloud forest. Mol Ecol 20:1829–1844. https://doi.org/10.1111/j.1365-294X.2011.05028.x
Cammack JA, Nelder MP (2010) Cool-weather activity of the forensically important hairy maggot blow fly Chrysomya rufifacies (Macquart) (Diptera: Calliphoridae) on carrion in upstate South Carolina, United States. Forensic Sci Int 195:139–142. https://doi.org/10.1016/j.forsciint.2009.12.007
Cardoso P, Barton PS, Birkhofer K, Chichorro F, Deacon C, Fartmann T, Fukushima CS, Gaigher R, Habel JC, Hallmann CA, Hill MJ, Hochkirch A, Kwak ML, Mammola S, Ari Noriega J, Orfinger AB, Pedraza F, Pryke JS, Roque FO, Settele J, Simaika JP, Stork NE, Suhling F, Vorster C, Samways MJ (2020) Scientists’ warning to humanity on insect extinctions. Biol Conserv. https://doi.org/10.1016/j.biocon.2020.108426
Chapman JW, Reynolds DR, Wilson K (2015) Long-range seasonal migration in insects: mechanisms, evolutionary drivers and ecological consequences. Ecol Lett 18:287–302. https://doi.org/10.1111/ele.12407
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes project analysis group (2011) The variant call format and vcftools. Bioinformatics. https://doi.org/10.1093/bioinformatics/btr330
Dawson BM, Barton PS, Wallman JF (2021) Field succession studies and casework can help to identify forensically useful Diptera. J Forensic Sci 66:2319–2328. https://doi.org/10.1111/1556-4029.14870
Dear JP (1985) Calliphoridae (Insecta: Diptera). Fauna of New Zealand 8.
Delnevo N, Piotti A, Carbognani M, van Etten EJ, Stock WD, Field DL, Byrne M (2021) Genetic and ecological consequences of recent habitat fragmentation in a narrow endemic plant species within an urban context. Biodiv and Conserv 30:3457–3478
Deschepper P, Todd TN, Virgilio M, De Meyer M, Barr NB, Ruiz-Arce R (2021) Looking at the big picture: Worldwide population structure and range expansion of the cosmopolitan pest Ceratitis capitata (Diptera, Tephritidae). Biol Invasions 23:3529–3543. https://doi.org/10.1007/s10530-021-02595-4
DiBlasi E, Johnson KP, Stringham SA, Hansen AN, Beach AB, Clayton DH, Bush SE (2018) Phoretic dispersal influences parasite population genetic structure. Mol Ecol 27:2770–2779. https://doi.org/10.1111/mec.14719
Dick CW, Etchelecu G, Austerlitz F (2003) Pollen dispersal of tropical trees (Dinizia excelsa: Fabaceae) by native insects and African honeybees in pristine and fragmented Amazonian rainforest. Mol Ecol 12:753–764. https://doi.org/10.1046/j.1365-294X.2003.01760.x
Drake VA, Gatehouse AG (1995) Insect migration: tracking resources through space and time. Cambridge University Press, United Kingdom
Dytham C, Shorrocks B (1992) Selection, patches and genetic variation: a cellular automaton modelling Drosophila populations. Evol Ecol 6:342–351. https://doi.org/10.1007/BF02270970
Eaton DAR, Overcast I (2020) Ipyrad: Interactive assembly and analysis of radseq datasets. Bioinformatics 36:2592–2594. https://doi.org/10.1093/bioinformatics/btz966
Eddy GW, Bushland R (1956) Screwworms that attack livestock. Yearb Agric 1956:172–175
Ellwood MDF, Foster WA (2004) Doubling the estimate of invertebrate biomass in a rainforest canopy. Nature 429:549–551. https://doi.org/10.1038/nature02560
Endersby NM, Hoffmann AA, McKechnie SW, Weeks AR (2007) Is there genetic structure in populations of Helicoverpa armigera from Australia? Entomol Exp Appl 122:253–263. https://doi.org/10.1111/j.1570-7458.2006.00515.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Ewers RM, Boyle MJW, Gleave RA, Plowman NS, Benedick S, Bernard H, Bishop TR, Bakhtiar EY, Chey VK, Chung AYC, Davies RG, Edwards DP, Eggleton P, Fayle TM, Hardwick SR, Homathevi R, Kitching RL, Khoo MS, Luke SH, March JJ, Nilus R, Pfeifer M, Rao SV, Sharp AC, Snaddon JL, Stork NE, Struebig MJ, Wearn OR, Yusah KM, Turner EC (2015) Logging cuts the functional importance of invertebrates in tropical rainforest. Nat Comm 6:6836. https://doi.org/10.1038/ncomms7836
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1093/genetics/131.2.479
Frichot E, François O (2015) LEA: An R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929. https://doi.org/10.1111/2041-210X.12382
Georges A, Gruber B, Pauly GB, White D, Adams M, Young MJ, Kilian A, Zhang X, Shaffer HB, Unmack PJ (2018) Genomewide SNP markers breathe new life into phylogeography and species delimitation for the problematic short-necked turtles (Chelidae: Emydura) of eastern Australia. Mol Ecol 27:5195–5213. https://doi.org/10.1111/mec.14925
Goudet J, Jombart T, Goudet MJ (2015) Package ‘hierfstat’. R package version 0.04‐22. http://github.com/jgx65/hierfstat.
Griffiths HM, Ashton LA, Walker AE, Hasan F, Evans TA, Eggleton P, Parr CL (2017) Ants are the major agents of resource removal from tropical rainforests. J Anim Ecol 87:293–300. https://doi.org/10.1111/1365-2656.12728
Gruber B, Unmack PJ, Berry OF, Georges A (2018) Dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Molecular Ecol Res 18:691–699. https://doi.org/10.1111/1755-0998.12745
Hoffmann AA, Sgrò CM, Kristensen TN (2017) Revisiting adaptive potential, population size, and conservation. Trends Ecol Evol 32:506–517. https://doi.org/10.1016/j.tree.2017.03.012
Hoffmann AA, White VL, Jasper M, Yagui H, Sinclair SJ, Kearney MR (2021) An endangered flightless grasshopper with strong genetic structure maintains population genetic variation despite extensive habitat loss. Ecol Evol 11:5364–5380. https://doi.org/10.1002/ece3.7428
Hogsette JA, Ruff JP (1985) Stable fly (Diptera: Muscidae) migration in northwest Florida. Environ Entomol 14:170–175. https://doi.org/10.1093/ee/14.2.170
Jabis MD, Ayers TJ, Allan GJ (2011) Pollinator-mediated gene flow fosters genetic variability in a narrow alpine endemic, Abronia alpina (Nyctaginaceae). Am J Bot 98:1583–1594. https://doi.org/10.3732/ajb.1000515
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jaya FR, Tanner JC, Whitehead MR, Doughty P, Keogh JS, Moritz CC, Catullo RA (2022) Population genomics and sexual signals support reproductive character displacement in Uperoleia (Anura: Myobatrachidae) in a contact zone. Mol Ecol. https://doi.org/10.1111/mec.16597
Jiang Y, Zhou L, Tucker CJ, Raghavendra A, Hua W, Liu YY, Joiner J (2019) Widespread increase of boreal summer dry season length over the congo rainforest. Nat Clim Chang 9:617–622. https://doi.org/10.1038/s41558-019-0512-y
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. https://doi.org/10.7717/peerj.281
Kavazos CRJ, Wallman JF (2012) Community composition of carrion-breeding blowflies (Diptera: Calliphoridae) along an urban gradient in south-eastern Australia. Landscape Urban Plan 106:183–190. https://doi.org/10.1016/j.landurbplan.2012.03.002
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) Diversity: An r package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kellermann VM, van Heerwaarden B (2019) Terrestrial insects and climate change: adaptive responses in key traits. Physiol Entomol 44:99–115. https://doi.org/10.1111/phen.12282
Kellermann VM, van Heerwaarden B, Hoffmann AA, Sgro CM (2006) Very low additive genetic variance and evolutionary potential in multiple populations of two rainforest Drosophila species. Evolution 60:1104–1108. https://doi.org/10.1111/j.0014-3820.2006.tb01187.x
Kelly M (2019) Adaptation to climate change through genetic accommodation and assimilation of plastic phenotypes. Philos T Roy Soc B 374:20180176. https://doi.org/10.1098/rstb.2018.0176
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89. https://doi.org/10.1007/978-1-61779-870-2_5
Kitching RL, Bergelson JM, Lowman MD, McIntyre S, Carruthers G (1993) The biodiversity of arthropods from Australian rainforest canopies: general introduction, methods, sites and ordinal results. Aust J Ecol 18:181–191. https://doi.org/10.1111/j.1442-9993.1993.tb00442.x
Kleinhans C, Willows-Munro S (2019) Low genetic diversity and shallow population structure in the endangered vulture. Gyps Coprotheres Sci Rep 9:5536. https://doi.org/10.1038/s41598-019-41755-4
Láruson ÁJ, Reed FA (2016) Stability of underdominant genetic polymorphisms in population networks. J Theor Biol 390:156–163
Laurance WF, Camargo JLC, Fearnside PM, Lovejoy TE, Williamson GB, Mesquita RCG, Meyer CFJ, Bobrowiec PED, Laurance SGW (2017) An Amazonian rainforest and its fragments as a laboratory of global change. Biol Rev 93:223–247. https://doi.org/10.1111/brv.12343
Legge S, Woinarski JCZ, Scheele BC, Garnett ST, Lintermans M, Nimmo DG, Whiterod NS, Southwell DM, Ehmke G, Buchan A, Gray J, Metcalfe DJ, Page M, Rumpff L, van Leeuwen S, Williams D, Ahyong ST, Chapple DG, Cowan M, Hossain MA, Kennard M, Macdonald S, Moore H, Marsh J, McCormack RB, Michael D, Mitchell N, Newell D, Raadik TA, Tingley R (2021) Rapid assessment of the biodiversity impacts of the 2019–2020 Australian megafires to guide urgent management intervention and recovery and lessons for other regions. Divers Distrib. https://doi.org/10.1111/ddi.13428
Lens L, Van Dongen S, Norris K, Githiru M, Matthysen E (2002) Avian persistence in fragmented rainforest. Science 298:1236–1238. https://doi.org/10.1126/science.1075664
Leung LKP, Dickman CR, Moore AL (1994) Genetic variation in fragmented populations of an Australian rainforest rodent, Melomys cervinipes. Pac Conserv Biol 1:58–65. https://doi.org/10.1071/PC930058
Linck E, Battey CJ (2019) Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol Ecol Resour 19:639–647. https://doi.org/10.1111/1755-0998.12995
Lloyd MW, Campbell L, Neel MC (2013) The power to detect recent fragmentation events using genetic differentiation methods. PLoS ONE. https://doi.org/10.1371/journal.pone.0063981
Mac Nally R, Bennett AF, Thomson JR, Radford JQ, Unmack G, Horrocks G, Vesk PA (2009) Collapse of an avifauna: climate change appears to exacerbate habitat loss and degradation. Div Distrib 15:720–730. https://doi.org/10.1111/j.1472-4642.2009.00578.x
Malloch JR (1927) Notes on Australian diptera. No XI Proc Lin Soc NSW 52:299–335
Mariani M, Fletcher MS, Haberle S, Chin H, Zawadzki A, Jacobsen G (2019) Climate change reduces resilience to fire in subalpine rainforests. Glob Change Biol 25:2030–2042. https://doi.org/10.1111/gcb.14609
Marsh J, Bal P, Fraser H, Umbers K, Greenville A, Rumpff L, Woinarski J (2021) Assessment of the impacts of the 2019–20 wildfires of southern and eastern Australia on invertebrate species. NESP Threatened species recovery hub project 8.3.1. Final report, Brisbane.
Martínez-Ramos M, Ortiz-Rodríguez IA, Piñero D, Dirzo R, Sarukhán J (2016) Anthropogenic disturbances jeopardize biodiversity conservation within tropical rainforest reserves. P Natl A Sci USA 113:5323–5328. https://doi.org/10.1073/pnas.1602893113
Melo-Carrillo A, Dunn JC, Cortés-Ortiz L (2020) Low genetic diversity and limited genetic structure across the range of the critically endangered Mexican howler monkey (Alouatta palliata mexicana). Am J Primatol. https://doi.org/10.1002/ajp.23160
Menken SBJ (1987) Is the extremely low heterozygosity level in Yponomeuta rorellus caused by bottlenecks? Evolution 41:630–637. https://doi.org/10.1111/j.1558-5646.1987.tb05834.x
Mesibov R (1998) Species-level comparison of litter invertebrates at two rainforest sites in Tasmania. Tasforests 10:141–153
Milá B, Wayne RK, Fitze P, Smith TB (2009) Divergence with gene flow and fine-scale phylogeographical structure in the wedge-billed woodcreeper, Glyphorynchus spirurus, a Neotropical rainforest bird. Mol Ecol 18:2979–2995. https://doi.org/10.1111/j.1365-294X.2009.04251.x
Moritz C, Hoskin C, Graham C, Hugall A, Moussalli A (2005) Historical biogeography, diversity and conservation of Australia’s tropical rainforest herpetofauna. In: Purvis A, Gittleman J, Brooks T (eds) Phylogeny and Conservation (Conservation Biology. Cambridge University Press, Cambridge
New TR (2018) Forests and insect conservation in Australia. Springer international publishing, Cham, Switzerland
Nolan RH, Boer MM, Collins L, Resco de Dios V, Clarke H, Jenkins M, Kenny B, Bradstock RA (2020) Causes and consequences of eastern Australia’s 2019–20 season of mega-fires. Glob Change Biol 26:1039–1041. https://doi.org/10.1111/gcb.14987
Norris KR (1959) The ecology of sheep blowflies in Australia. In A. Keast, Crocker R. L. Christian, C. S. (Eds) Biogeography and Ecology in Australia. Dordrecht, Springer Netherlands: 514–544
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara RB, Simpson GL, Solymos P, Stevens MH, Wagner H (2013) Package ‘vegan’. Community ecology package, version 2.5–7 https://CRAN.R-project.org/package=vegan
Olson DM (1994) The distribution of leaf litter invertebrates along a Neotropical altitudinal gradient. J Trop Ecol 10:129–150. https://doi.org/10.1017/S0266467400007793
Pembleton L, Cogan N, Forster J (2013) StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol Ecol Res 13:946–952. https://doi.org/10.1111/1755-0998.12129
Pfeiler E, Markow TA (2017) Population connectivity and genetic diversity in long-distance migrating insects: divergent patterns in representative butterflies and dragonflies. Biol J Lin Soc 122:479–486. https://doi.org/10.1093/biolinnean/blx074
Popa-Báez ÁD, Catullo R, Lee SF, Yeap HL, Mourant RG, Frommer M, Sved JA, Cameron EC, Edwards OR, Taylor PW, Oakeshott JG (2020) Genome-wide patterns of differentiation over space and time in the Queensland fruit fly. Sci Rep 10:10788. https://doi.org/10.1038/s41598-020-67397-5
Qin Y, Krosch MN, Schutze MK, Zhang Y, Wang X, Prabhakar CS, Susanto A, Hee AKW, Ekesi S, Badji K, Khan M, Wu J, Wang Q, Yan G, Zhu L, Zhao Z, Liu L, Clarke AR, Li Z (2018) Population structure of a global agricultural invasive pest, Bactrocera dorsalis (Diptera: Tephritidae). Evol Appl 11:1990–2003. https://doi.org/10.1111/eva.12701
R Core Team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.Rproject.org/
Radespiel U, Bruford MW (2014) Fragmentation genetics of rainforest animals: insights from recent studies. Conserv Genet 15:245–260. https://doi.org/10.1007/s10592-013-0550-3
Razgour O, Forester B, Taggart JB, Bekaert M, Juste J, Ibáñez C, Puechmaille SJ, Novella-Fernandez R, Alberdi A, Manel S (2019) Considering adaptive genetic variation in climate change vulnerability assessment reduces species range loss projections. P of Natl A Sci USA 116:10418. https://doi.org/10.1073/pnas.1820663116
Robertson EP, Fletcher RJ, Austin JD (2018) Microsatellite polymorphism in the endangered snail kite reveals a panmictic, low diversity population. Conserv Genet 19:337–348. https://doi.org/10.1007/s10592-017-1003-1
Ruiz-Carbayo H, Espelta JM, Pino J, Hampe A, Bonal R (2022) Contrasting genetic population structures in acorn weevils (Curculio spp) in expanding forests: the effects of differences in resource-tracking strategies. Insect Conserv Diver. https://doi.org/10.1111/icad.12603
Sadanandan KR, Rheindt FE (2015) Genetic diversity of a tropical rainforest understory bird in an urban fragmented landscape. The Condor 117:447–459. https://doi.org/10.1650/CONDOR-14-199.1
Samways MJ, Barton PS, Birkhofer K, Chichorro F, Deacon C, Fartmann T, Fukushima CS, Gaigher R, Habel JC, Hallmann CA, Hill MJ, Hochkirch A, Kaila L, Kwak ML, Maes D, Mammola S, Noriega JA, Orfinger AB, Pedraza F, Pryke JS, Roque FO, Settele J, Simaika JP, Stork NE, Suhling F, Vorster C, Cardoso P (2020) Solutions for humanity on how to conserve insects. Biol Conserv. https://doi.org/10.1016/j.biocon.2020.108427
Satterfield DA, Sillett TS, Chapman JW, Altizer S, Marra PP (2020) Seasonal insect migrations: massive, influential, and overlooked. Front Ecol Environ 18:335–344. https://doi.org/10.1002/fee.2217
Saunders DS, Hayward SA (1998) Geographical and diapause-related cold tolerance in the blow fly, Calliphora vicina. J Insect Physiol 44:541–551. https://doi.org/10.1016/s0022-1910(98)00049-3
Schmidt TL, Jasper ME, Weeks AR, Hoffmann AA (2021) Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol Evol 12:1888–1898. https://doi.org/10.1111/2041-210x.13659
Shapcott A (2000) Conservation and genetics in the fragmented monsoon rainforest in the Northern Territory, Australia: a case study of three frugivore-dispersed species. Aust J Bot 48:397–407. https://doi.org/10.1071/BT98081
Shoo LP, Williams SE, Hero JM (2005) Climate warming and the rainforest birds of the Australian wet tropics: using abundance data as a sensitive predictor of change in total population size. Biol Conserv 125:335–343. https://doi.org/10.1016/j.biocon.2005.04.003
Snaddon JL, Turner EC, Foster WA (2008) Children’s perceptions of rainforest biodiversity: which animals have the lion’s share of environmental awareness? PLoS ONE. https://doi.org/10.1371/journal.pone.0002579
Sontigun N, Sukontason KL, Klong-klaew T, Sanit S, Samerjai C, Somboon P, Thanapornpoonpong SN, Amendt J, Sukontason K (2018) Bionomics of the oriental latrine fly Chrysomya megacephala (Fabricius) (Diptera: Calliphoridae): temporal fluctuation and reproductive potential. Parasite Vector 11:415. https://doi.org/10.1186/s13071-018-2986-2
Stork NE, Grimbacher PS (2006) Beetle assemblages from an Australian tropical rainforest show that the canopy and the ground strata contribute equally to biodiversity. P R Soc B 273:1969–1975. https://doi.org/10.1098/rspb.2006.3521
Su J, Yan Y, Song J, Li J, Mao J, Wang N, Wang W, Du FK (2018) Recent fragmentation may not alter genetic patterns in endangered long-lived species: evidence from Taxus cuspidata. Front Plant Sci 9:1571. https://doi.org/10.3389/fpls.2018.01571
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D (2016) Directional genetic differentiation and relative migration. Ecol Evol 6:3461–3475. https://doi.org/10.1002/ece3.2096
Szpila K, Wallman JF (2016) Morphology and identification of first instar larvae of Australian blowflies of the genus Chrysomya of forensic importance. Acta Trop 162:146–154. https://doi.org/10.1016/j.actatropica.2016.06.006
Taylor GS, Braby MF, Moir ML, Harvey MS, Sands DPA, New TR, Kitching RL, McQuillan PB, Hogendoorn K, Glatz RV, Andren M, Cook JM, Henry SC, Valenzuela I, Weinstein P (2018) Strategic national approach for improving the conservation management of insects and allied invertebrates in Australia. Aust Entomol 57:124–149. https://doi.org/10.1111/aen.12343
Thioulouse J, Dray S, Dufour A, Siberchicot A, Jombart T, Pavoine S (2018) Multivariate analysis of ecological data with ade4. Springer, New York, NY
Trouvé R, Oborne L, Baker PJ (2021) The effect of species, size, and fire intensity on tree mortality within a catastrophic bushfire complex. Ecol Appl. https://doi.org/10.1002/eap.2383
Tsuda Y, Hayashi T, Higa Y, Hoshino K, Kasai S, Tomita T, Kurahashi H, Kobayashi M (2009) Dispersal of a blow fly, Calliphora nigribarbis, in relation to the dissemination of highly pathogenic avian influenza virus. Jpn J Infect Dis 62:294–297
Vinogradova EB, Reznik SY (2013) Induction of larval diapause in the blowfly, Calliphora vicina R.-D. (Diptera, Calliphoridae) under field and laboratory conditions. Entomol Rev 93:935–941. https://doi.org/10.1134/S0013873813080010
Wallman JF (2002) A key to the adults of species of blowflies in southern Australia known or suspected to breed in carrion. Med Vet Entomol 15:433–437. https://doi.org/10.1046/j.0269-283x.2001.00331.x
Wang X, Yang X, Zhou L, Wyckhuys KAG, Jiang S, Van Liem N, Vi LX, Ali A, Wu K (2021) Population genetics unveils large-scale migration dynamics and population turnover of Spodoptera exigua. Pest Manag Sci 78:612–625. https://doi.org/10.1002/ps.6670
Wardhaugh CW, Stork NE, Edwards W, Grimbacher PS (2012) The overlooked biodiversity of flower-visiting invertebrates. PLoS ONE. https://doi.org/10.1371/journal.pone.0045796
Weir BS, Goudet J (2017) A unified characterization of population structure and relatedness. Genetics 206:2085–2103. https://doi.org/10.1534/genetics.116.198424
Wilke ABB, Wilk-da-Silva R, Marrelli MT (2017) Microgeographic population structuring of Aedes aegypti (Diptera: Culicidae). PLoS ONE. https://doi.org/10.1371/journal.pone.0185150
Williams SE, Bolitho EE, Fox S (2003) Climate change in Australian tropical rainforests: an impending environmental catastrophe. P R Soc B 270:1887–1892. https://doi.org/10.1098/rspb.2003.2464
Woltmann S, Kreiser BR, Sherry TW (2012) Fine-scale genetic population structure of an understory rainforest bird in Costa Rica. Conserv Genet 13:925–935. https://doi.org/10.1007/s10592-012-0341-2
Wright BR, Grueber CE, Lott MJ, Belov K, Johnson RN, Hogg CJ (2019) Impact of reduced-representation sequencing protocols on detecting population structure in a threatened marsupial. Mol Biol Rep 46:5575–5580. https://doi.org/10.1007/s11033-019-04966-6
Yeates D, Harvey M, Austin A (2003) New estimates for terrestrial arthropod species-richness in Australia. Records South Australian Museum Monograph Series 7:231–241
Yu G (2020) Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics. https://doi.org/10.1002/cpbi.96
Acknowledgements
The scientific licence number associated with this project is SL101850. All applicable institutional and/or national guidelines for the care and use of animals were followed. The authors thank the Paddy Pallin Foundation and the Royal Zoological Society of New South Wales for their financial support. We thank Finlay Davidson for his substantial assistance with field work in southern NSW.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This research was supported by the Paddy Pallin Foundation and the Royal Zoological Society of New South Wales.
Author information
Authors and Affiliations
Contributions
NJB: Designed and performed research, analysed data, and wrote the manuscript. JFW: Helped conceptualise the project, provided funding, and edited the manuscript. NPJ: Assisted with fieldwork, sample preparation, analysis, and edited the manuscript. BMD: Assisted with fieldwork, designed the blowfly field bait, and edited the manuscript. JSH: Conducted spatial analysis of rainforest fragmentation, designed figures, and edited the manuscript. AM: Helped design the research project, guided the research and data analysis, and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Additional information
Communicated by Kyle J Haynes.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Butterworth, N.J., Wallman, J.F., Johnston, N.P. et al. The blowfly Chrysomya latifrons inhabits fragmented rainforests, but shows no population structure. Oecologia 201, 703–719 (2023). https://doi.org/10.1007/s00442-023-05333-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00442-023-05333-w